The Quantum Insider (TQI) is the leading online resource dedicated exclusively to Quantum Computing.




If Spider-Man were real, he could help scientists revolutionize healthcare. Since he’s not, researchers develop creative ways to produce spider silk to study.

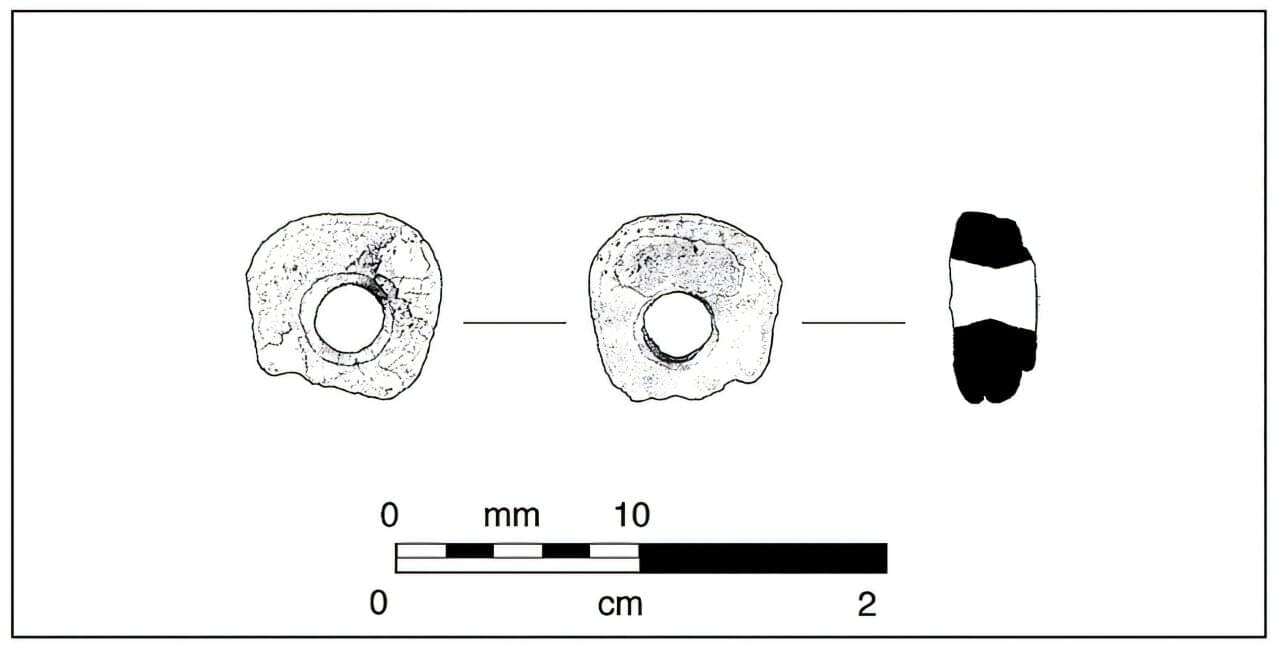
Archaeologists have discovered two giant rock scallop beads that push back the clock on the first appearance of these high-status ornaments. The discovery shows that these prized marine beads were traded hundreds of miles from the California coast thousands of years earlier than previously thought.
Radiocarbon dating places one of the beads at roughly 8,500 years old, making it the oldest ever discovered. More unusually, they were found more than 42 miles (68 kilometers) from the nearest coast, extending the known range of these ornaments from the Channel Islands to the California interior.
“It’s cool that this is the oldest of its kind identified so far, but cooler still to view it as part of a broader Early Holocene cultural landscape seemingly well-established in this locale as much as 10,000 years ago,” said Barry A. Price, an archaeologist at Applied EarthWorks Inc. and one of the study’s authors, along with Simone Schinsing and Jasmine Kidwell. The study is published in California Archaeology.

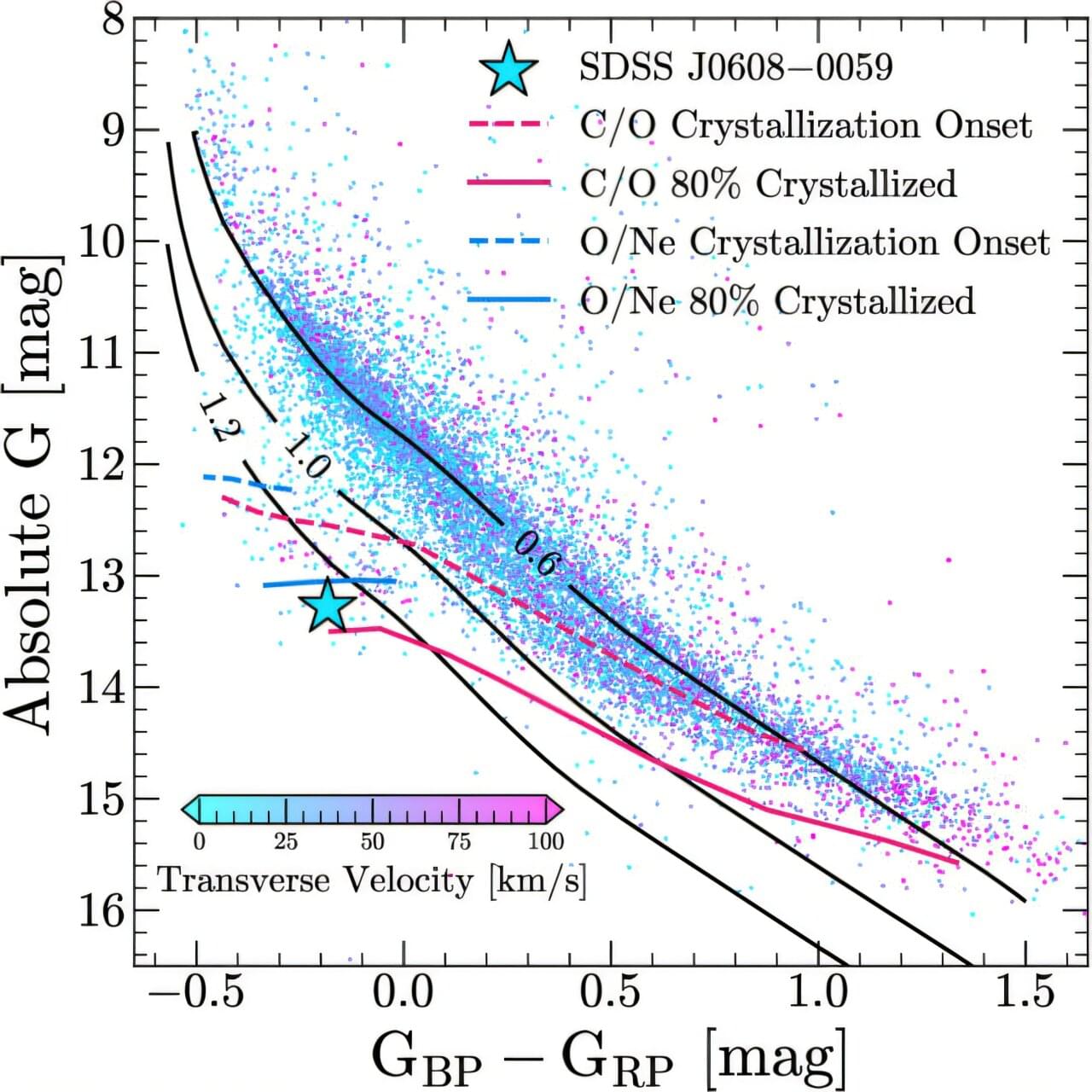
Astronomers have found evidence that one of the most massive white dwarfs known has an oxygen-neon core instead of the more common carbon-oxygen core. The finding is important because the composition of a white dwarf’s core determines how it will evolve. A paper outlining this discovery was published in The Astrophysical Journal.
Typically, white dwarfs have a mass of 0.5–0.7 times the sun’s mass. Such objects have a core made up mainly of carbon and oxygen (C/O core). When they have stellar companions, these dense objects can accumulate matter from them and eventually produce a Type Ia supernova. Ultramassive white dwarfs, with masses above roughly 1.05–1.1 times the sun’s mass, tell a different story that is not yet fully understood.
These more massive white dwarfs are thought to form from “ancestor” or progenitor stars in the range of about 8–10 times the sun’s mass. In these heavier progenitors, the core reaches higher temperatures and densities, allowing carbon to ignite and fuse further into oxygen and neon (O/Ne core). This does not happen in the cores of lower-mass stars, which stop fusing once they have built up carbon and oxygen, lacking the required core temperatures.
{kind=link}