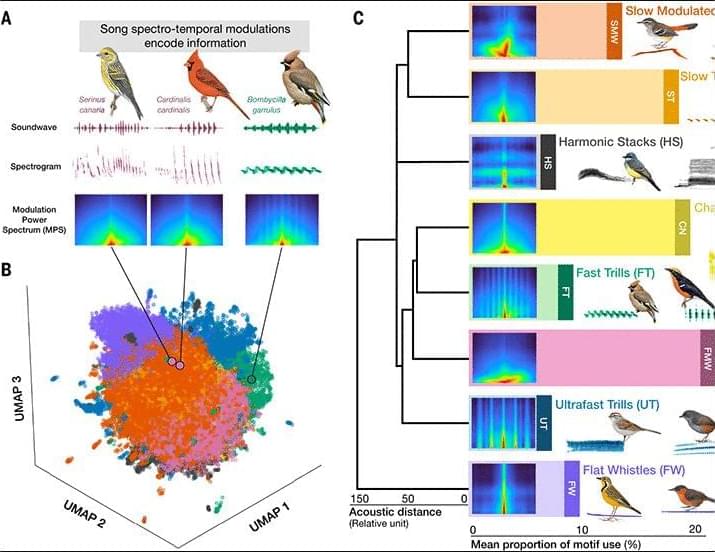
Although bird songs are classic models for understanding the evolution of vocal communication, their global diversity has long made the development of a unifying framework challenging. By analyzing the acoustic architecture of songs from more than 3,000 passerine species worldwide, we show that this acoustic space can be structured around eight elemental motifs. The differential use of these motifs is driven by a combination of species’ biological traits (social organization, morphology, and mating system) and the physics of sound propagation. In tropical rainforests, environmental filtering for transmission efficiency favors structurally simple motifs, such as flat whistles.