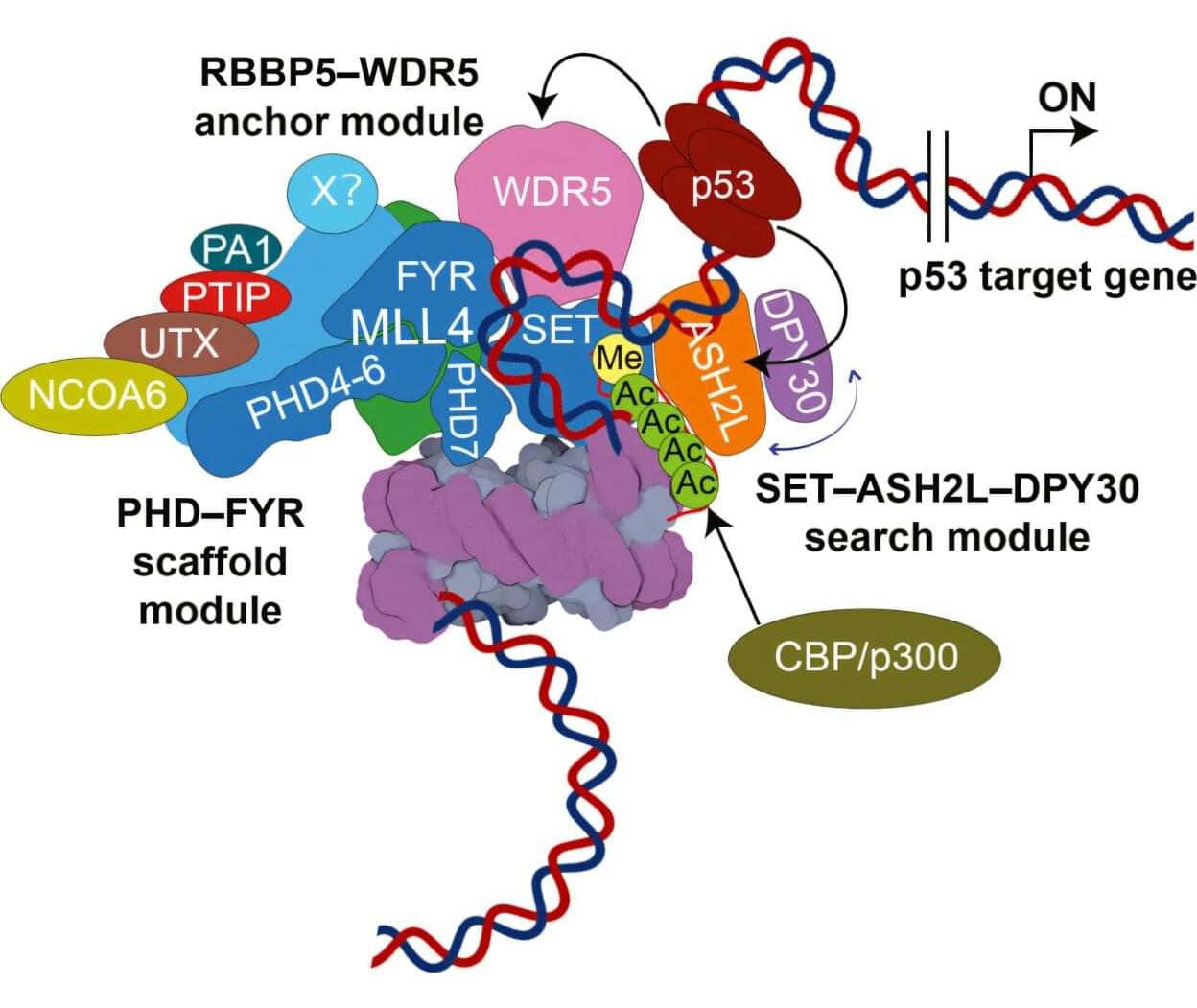
The combination drug ceftazidime-avibactam (CZA) is a last line of defense against the common Pseudomonas aeruginosa hospital bug: It’s the drug that gets called in when nothing else works, but there’s now evidence that it may not keep working for long.
Based on an analysis of two critically ill patients with P. aeruginosa infections, the bacteria are developin g genetic mutations that change the enzymes they produce – and can ward off an attack from CZA.
Researchers led by a team from Tongji University in China have now published a new paper in Microbiology Spectrum detailing the mutations and what it might mean for fighting P. aeruginosa in the future.