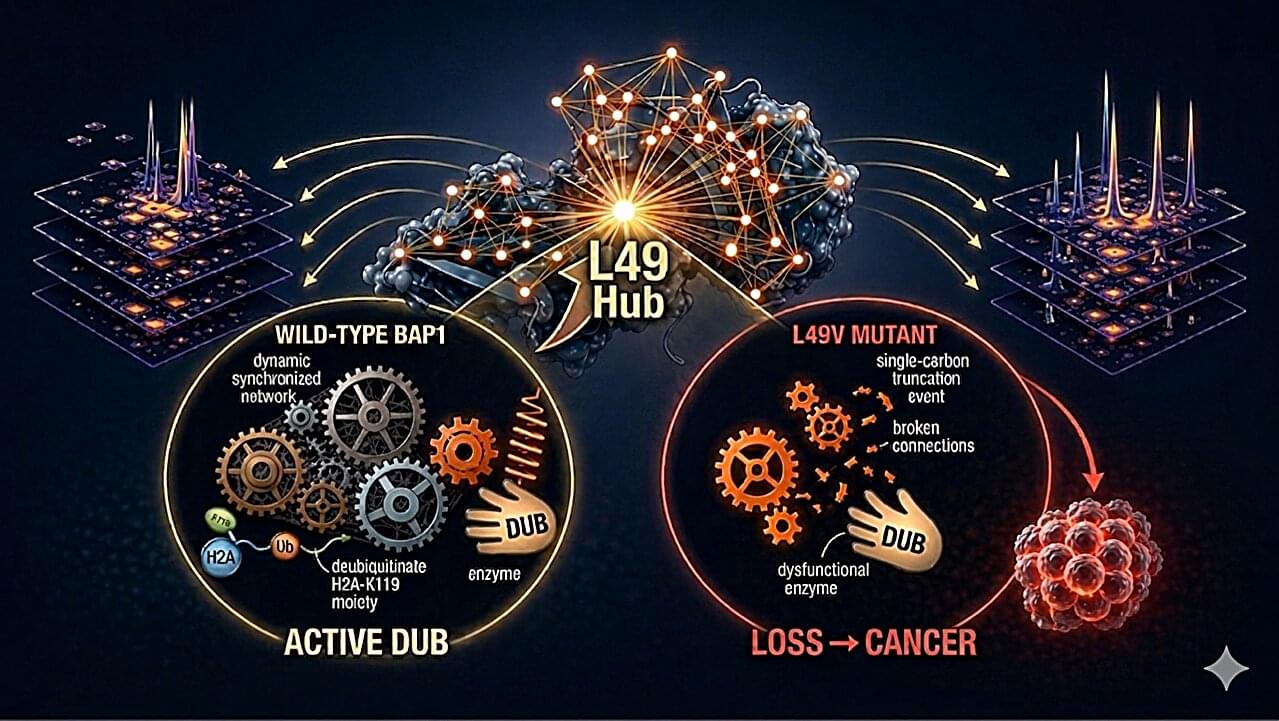
Scientists at the Institute of Biochemical Sciences at National Taiwan University have uncovered how tiny genetic changes can disable one of the body’s most important tumor-suppressing proteins. Their study, published in Nature Communications, reveals how cancer-associated mutations interfere with the function of BRCA1-associated protein 1 (BAP1), a protein that helps maintain normal cell growth and is frequently mutated in cancers such as mesothelioma, uveal melanoma and kidney cancer.
Although many cancer mutations in BAP1 have been identified over the years, it has remained unclear exactly how they impair the protein. To answer this question, the research team examined nearly 50 cancer-associated mutations using advanced nuclear magnetic resonance (NMR) spectroscopy, computer simulations and biochemical experiments.