LeCun and Oriol Vinyals join 224 Ventures, a new AI fund with $100M+ in assets. It targets early stage AI startups with $1M-$5M checks. LPs are mostly AI insiders.





If Spider-Man were real, he could help scientists revolutionize healthcare. Since he’s not, researchers develop creative ways to produce spider silk to study.

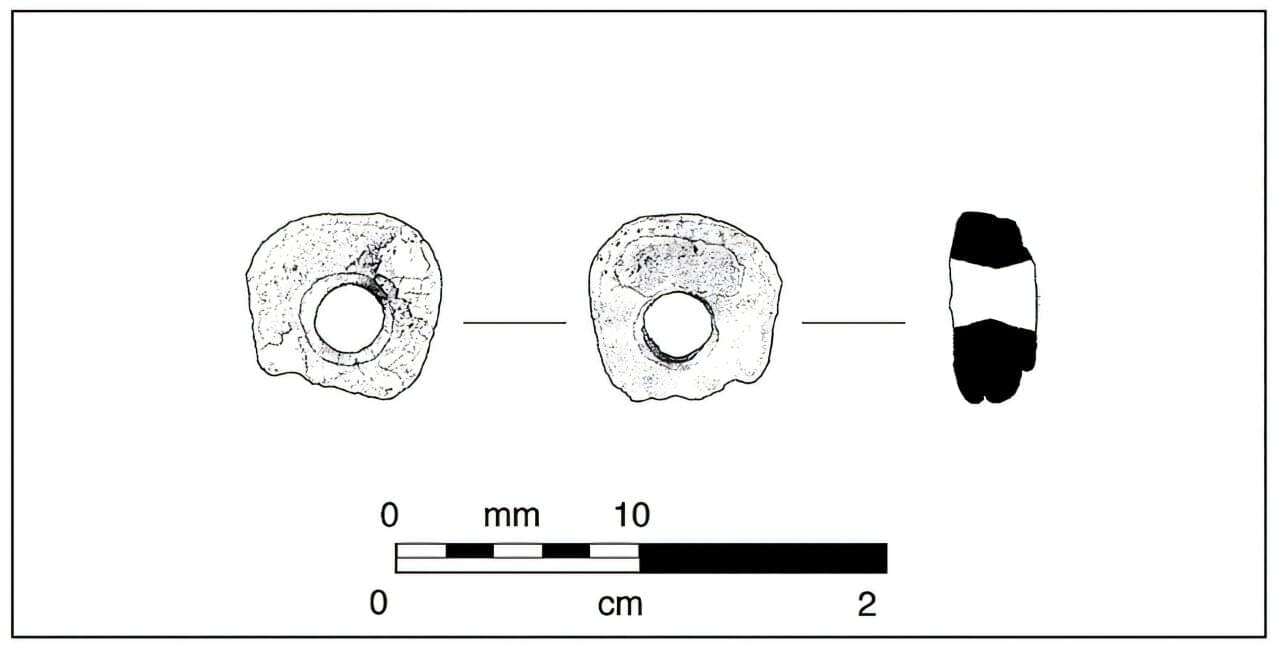
Archaeologists have discovered two giant rock scallop beads that push back the clock on the first appearance of these high-status ornaments. The discovery shows that these prized marine beads were traded hundreds of miles from the California coast thousands of years earlier than previously thought.
Radiocarbon dating places one of the beads at roughly 8,500 years old, making it the oldest ever discovered. More unusually, they were found more than 42 miles (68 kilometers) from the nearest coast, extending the known range of these ornaments from the Channel Islands to the California interior.
“It’s cool that this is the oldest of its kind identified so far, but cooler still to view it as part of a broader Early Holocene cultural landscape seemingly well-established in this locale as much as 10,000 years ago,” said Barry A. Price, an archaeologist at Applied EarthWorks Inc. and one of the study’s authors, along with Simone Schinsing and Jasmine Kidwell. The study is published in California Archaeology.
{kind=link}
{kind=link}