Our brains are large compared with other animals, so it is tempting to assume there was an evolutionary advantage to them – but that may not be true at all

What happens when the laws of the universe turn against existence itself?
In this cinematic 4K documentary, we journey through a hierarchy of cosmic catastrophes that challenge the survival of any civilization, from the localized death of planets to the absolute collapse of physical laws.
▶A Film by: Scienshell Studio.
In a universe governed by deep time and immense forces, disaster is not a matter of if, but when. From worlds frozen in perpetual schizophrenia by tidal locking to the ultimate recollapse of space-time, civilizations must either evolve to engineer the cosmos or face total eradication.
But as the scale of destruction grows, the line between technology and natural law begins to blur. For those who survive the end of the universe, reality itself becomes a blank canvas.
In this video, you’ll discover:
00:00 Introduction.
02:35 Tidally Locked Worlds and Planetary Accelerators.
05:35 Stellar Storms and Planetary Shield.
08:43 Supernovae and Star Lifting.
11:53 Supermassive Black Holes and The Space Nomads.
14:54 Interstellar Interceptors: The Predator and the Prey.
17:08 The Inflaton Field and The Big Crunch.
21:01 Higgs Field Decay: The Death of Matter and Gravity.
22:31 Beyond Physics: Civillization with godly powers.
▶ About This Video.
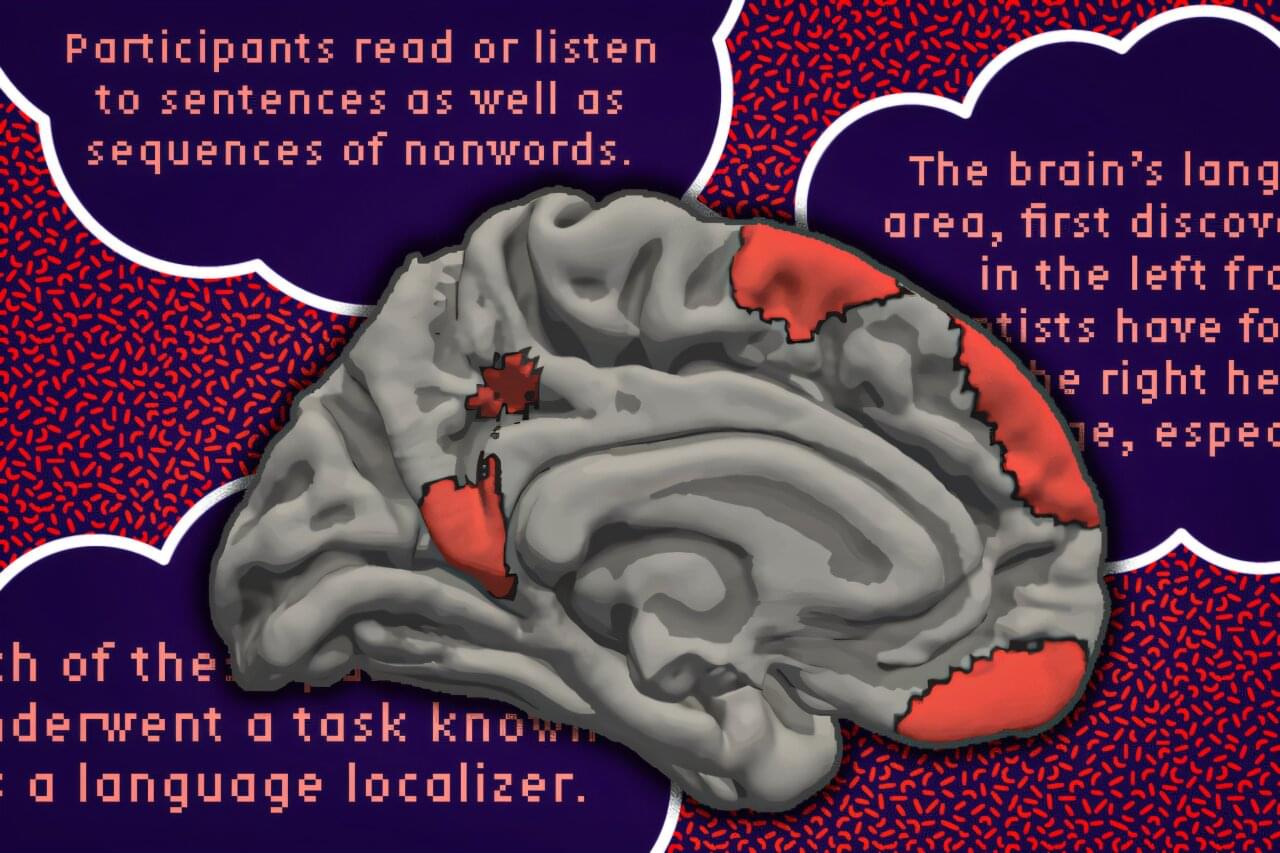
For decades, neuroscientists have known that specific regions in the brain’s left hemisphere are responsible for processing language. However, a new study by MIT researchers shows that language processing also occurs in many other parts of the brain.
Using functional magnetic resonance imaging (fMRI) data from more than 700 people, the researchers identified 17 additional regions of the brain that appear to play a role in language. These regions are scattered across the brain, including parts of the cerebellum, hippocampus and cerebral cortex, and they make up about 5% of the total volume of the adult brain—about the size of a large strawberry.
“Even though there are all these distant components, it’s pretty restricted in terms of volume. You don’t need that much of the brain to do language,” says Evelina Fedorenko, an MIT associate professor of brain and cognitive sciences, a member of MIT’s McGovern Institute for Brain Research, and the senior author of the study.
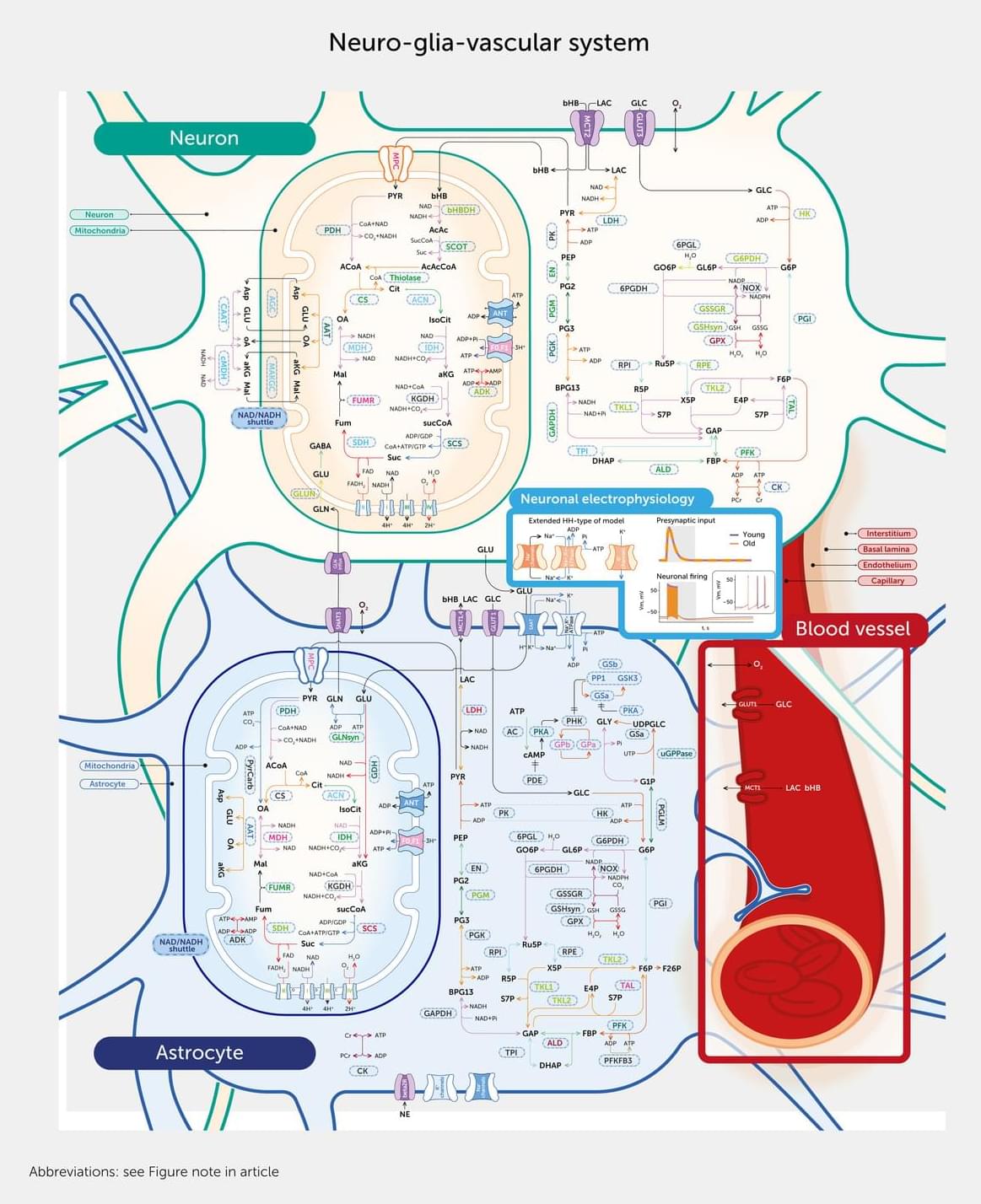
Age-related neurodegenerative disorders, including dementia, are a major global health concern. This article describes the first comprehensive, data-driven molecular model of the neuro-glia-vascular system to explore the complex relationships between the aging brain, energy metabolism, blood flow, and neuronal activity. Comprising 16,800 interaction pathways, the model includes all key enzymes, transporters, metabolites, and circulatory factors vital for neuronal electrical activity. We found significant alterations in metabolite concentrations and differential effects on adenosine triphosphate (ATP) supply in neurons and astrocytes and within subcellular compartments in aged brains and identified reduced sodium/potassium adenosine triphosphatase (Na+/K+-ATPase) activity as the leading cause of impaired neuronal action potentials.
On Sunday, July 5, 2026, at 1 p.m. U.S. Pacific Time, watch a compilation stream of four additional presentations from the May 2–3, 2026, sessions at the University of California, Berkeley Conference on Aging and Longevity (BerkeleyCAL), hosted by Professor Steven A. Garan, Director of Bioinformatics at the Center for Research and Education on Aging.
These presentations focus key insights in geroscience, both from its history and in regard to promising future directions and some implications for effective advocacy; they are delivered by some of the leading researchers in longevity science – Aubrey de Grey, Brendan Hughes, Felipe Sierra, and Michael West. Three of the presentations include question-and-answer sessions.
Dr. Aubrey de Grey of the Longevity Escape Velocity Foundation (LEVF) discusses the historical approaches to viewing aging and their shortcomings, as well as the damage-repair approach that he has championed and its prospects for rejuvenating the body. He also discusses implications for advocacy and which tactics could be more effective in bringing the public on board. Note: This presentation is an excerpt, captured by USTP Chairman Stolyarov on his phone camera. It is being made available due to the official recording having been lost.
Dr. Brendan Hughes from the Buck Institute discusses his thesis research on how DNA damage shapes unique, disease-relevant senescent cell states in neurons and other brain cell types. He details a methodology involving the direct differentiation of fibroblasts into neurons and oligodendrocytes to better understand aging-related cellular responses and potential therapeutic targets for Alzheimer’s disease. Dr. Hughes also highlights the importance of basic research in developing future interventions, such as senescence-targeted therapies or DNA repair modulations. The question-and-answer session includes a question from USTP Chairman Stolyarov to Dr. Hughes.
Dr. Felipe Sierra advocates for a shift in geroscience from solely targeting age-related diseases to focusing on maintaining intrinsic health and functional capacity. He proposes that molecular resilience acts as the crucial link between aging biology and long-term health, suggesting that strengthening this resilience could prevent the onset of multiple morbidities. Ultimately, he calls for more robust longitudinal studies and clinical trials that prioritize health-span metrics over the traditional, disease-centered approach to geriatric medicine.
Dr. Michael West explores the biological dichotomy between mortal somatic cells and the immortal germline to explain the fundamental mechanisms of aging and cellular regeneration. He discusses the history of stem-cell research and his work on telomeres and nuclear transfer, which demonstrated that developmental aging and cellular differentiation are reversible processes. Dr. West proposes a new approach to regenerative medicine that focuses on unlocking the body’s innate potential by targeting heterochrony genes to combat chronic degenerative diseases.
In principle, anyone denying the existence of some type of thing is an eliminativist with regard to that type of thing. Thus, there have been a number of eliminativists about different aspects of human nature in the history of philosophy. For example, hard determinists like Holbach (1770) are eliminativists with regard to free will because they claim there is no dimension of human psychology that corresponds to our commonsense notion of freedom. Similarly, by denying that there is an ego or persisting subject of experience, Hume (1739) was arguably an eliminativist about the self. Reductive materialists can be viewed as eliminativists with respect to an immaterial soul.
Nevertheless, contemporary eliminative materialism—the sort of eliminativism that denies the existence of specific types of mental states—is a relatively new theory with a very short history. The term was first introduced by James Cornman in a 1968 article entitled “On the Elimination of ‘Sensations’ and Sensations” (Cornman, 1968). However, the basic idea goes back at least as far as C.D. Broad’s classic, The Mind and its Place in Nature (Broad, 1925). Here Broad discusses, and quickly rejects, a type of “pure materialism” that treats mental states as attributes that apply to nothing in the world (pp. 607–611). Like many future writers (see section 4.1 below), Broad argued that such a view is self-contradictory since it (presumably) presupposes the reality of misjudgments which are themselves a type of mental state.
Apart from Broad’s discussion, the main roots of eliminative materialism can be found in the writings of a number of mid-20th century philosophers, most notably Wilfred Sellars, W.V.O. Quine, Paul Feyerabend, and Richard Rorty. In his important 1956 article, “Empiricism and the Philosophy of Mind”, Sellars introduced the idea that our conception of mentality may be derived not from direct access to the inner workings of our own minds, but instead from a primitive theoretical framework that we inherit from our culture. While Sellars himself regarded this theoretical framework as empirically correct, his claim that our conception of the mind is theory-based, and at least in principle falsifiable, would be influential to later supporters of eliminativism.

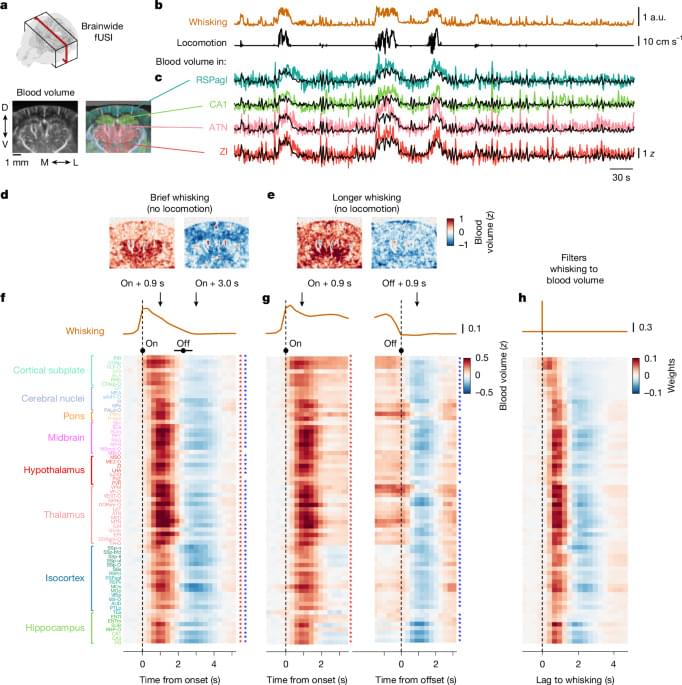
An interesting new approach to more accurately predicting blood flow in the mouse brain based on the activity of neurons correlated positively or negatively with arousal (as measured by whisking). Neuropixels and functional ultrasound imaging were used to simultaneously record from neurons and map blood flow, allowing the authors to derive their model.
Combined functional ultrasound imaging and Neuropixels recording of mouse brains identify two neuronal populations with opposing arousal-related activity and distinct haemodynamic response functions, that occur throughout the brain.
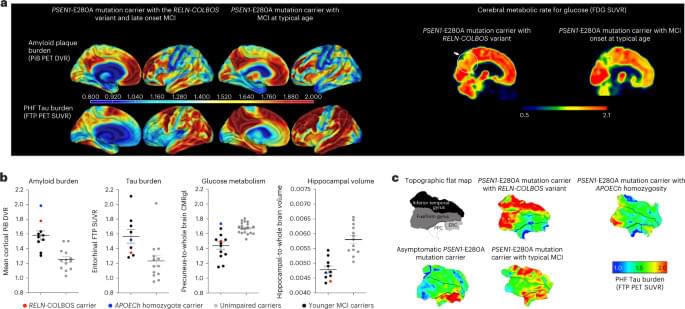
Fascinating case study on the neuroprotective effects of a mutant reelin allele (RELN–COLBOS) which delayed disease progression in a patient with autosomal dominant Alzheimer’s disease (ADAD). A promising therapeutic target!
Case report of an individual heterozygous for a rare RELN–COLBOS variant that confers resilience, via a gain-of-function mechanism, to Alzheimer’s disease.