2 Curriculum in Genetics and Molecular Biology.
3Department of Biochemistry and Biophysics, and.
4Department of Radiation Oncology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA.
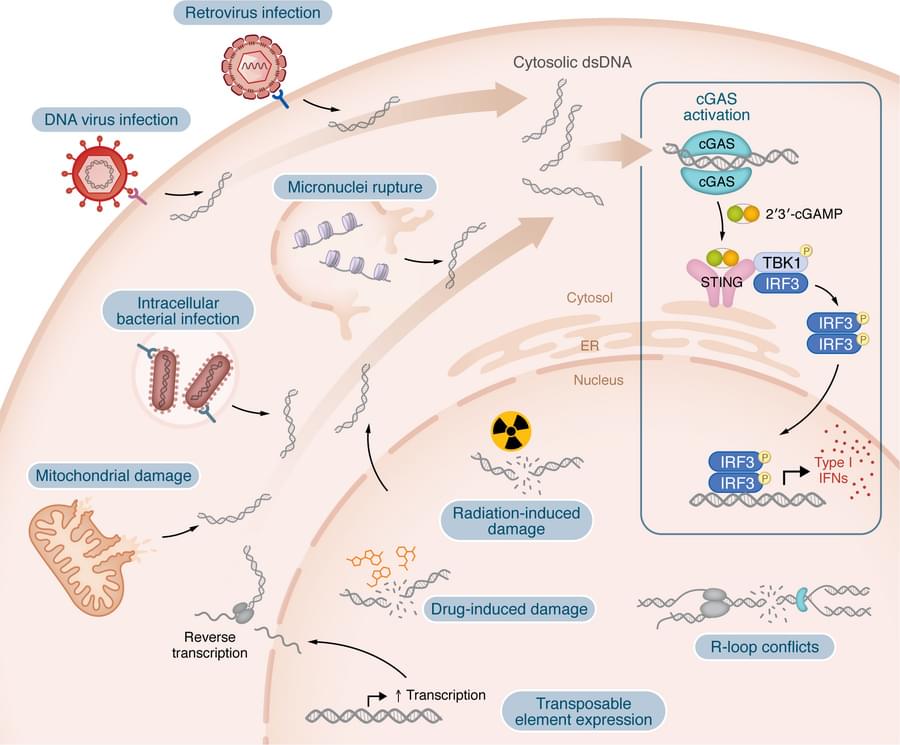

Human trisomy 21, responsible for Down syndrome, is the most prevalent genetic cause of cognitive impairment and remains a key focus for prenatal and preimplantation diagnosis. However, research directed toward eliminating supernumerary chromosomes from trisomic cells is limited. The present study demonstrates that allele-specific multiple chromosome cleavage by clustered regularly interspaced palindromic repeats Cas9 can achieve trisomy rescue by eliminating the target chromosome from human trisomy 21 induced pluripotent stem cells and fibroblasts. Unlike previously reported allele-nonspecific strategies, we have developed a comprehensive allele-specific (AS) Cas9 target sequence extraction method that efficiently removes the target chromosome. The temporary knockdown of DNA damage response genes increases the chromosome loss rate, while chromosomal rescue reversibly restores gene signatures and ameliorates cellular phenotypes. Additionally, this strategy proves effective in differentiated, nondividing cells. We anticipate that an AS approach will lay the groundwork for more sophisticated medical interventions targeting trisomy 21.
Keywords: CRISPR/Cas; Down syndrome; allele specificity; chromosome cut; chromosome loss; human trisomy 21.
© The Author(s) 2025. Published by Oxford University Press on behalf of National Academy of Sciences.
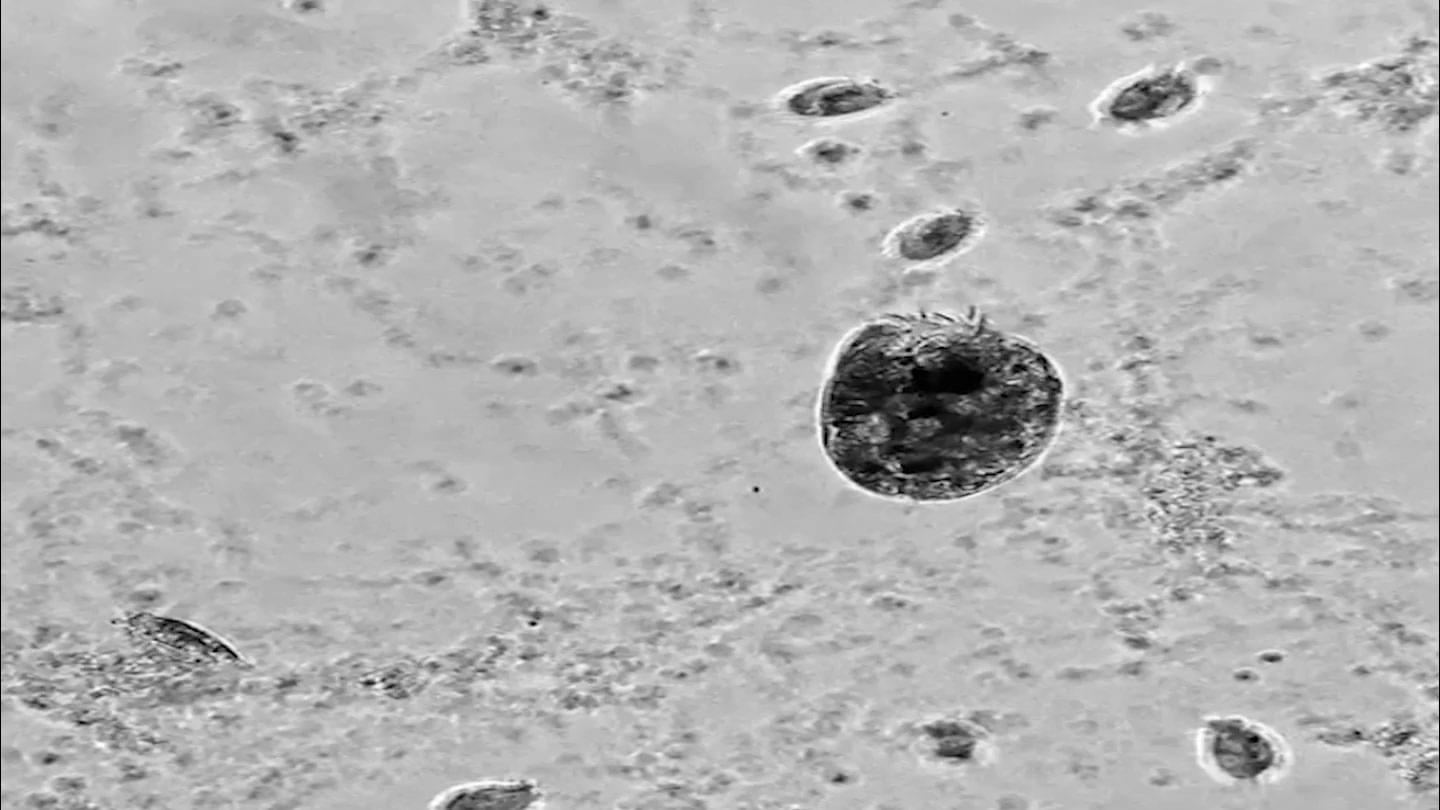
A newly discovered microbe is like a mini version of the Hulk.
Euplotes gigatrox is a single-celled protist that resembles an insect. It grazes on bacteria and other tiny microbes. Sometimes a small number of the protists balloon into “supergiants” more than twice their regular size. The huge cells cannibalize their smaller, genetically identical brethren. The triggers for the change aren’t entirely clear, but it tends to happen when there is plenty of food, researchers reported May 14 in the Proceedings of the National Academy of Sciences.
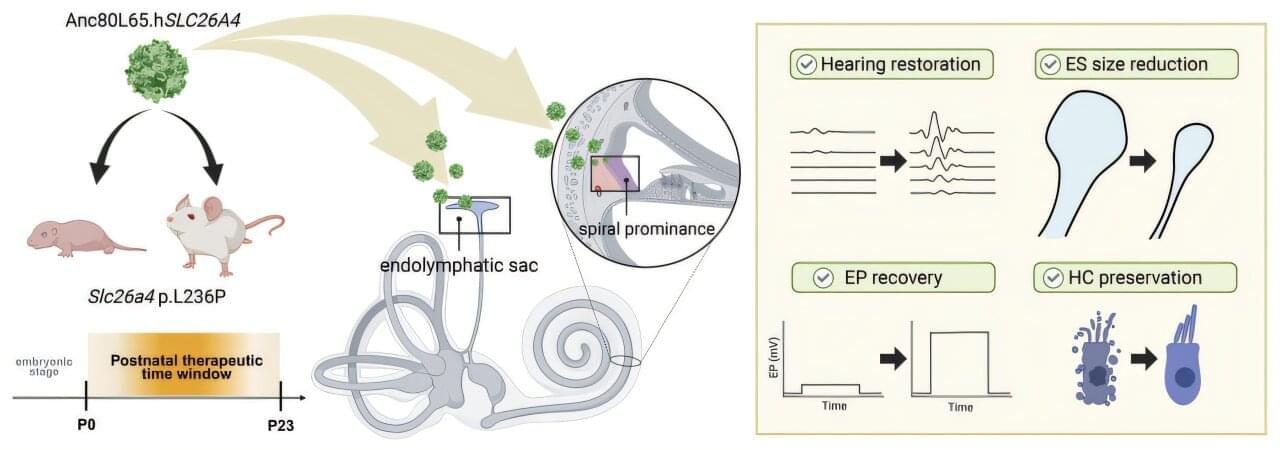
Hereditary hearing loss affects millions globally, with mutations in the SLC26A4 gene among the most common genetic triggers, particularly across Asian populations. This condition leads to severe-to-profound deafness accompanied by inner ear malformations, such as an abnormally enlarged vestibular aqueduct and endolymphatic sac.
While gene replacement therapies have long held immense potential, experimental interventions have historically been restricted to the embryonic stage. Delivering genetic material before birth presents steep ethical and practical hurdles, creating a critical roadblock for real-world medical applications.
A new study by researchers at the Icahn School of Medicine at Mount Sinai overturns a longstanding assumption about how mRNA vaccines generate immunity, revealing that certain non-immune cells help determine vaccine effectiveness.
The study, published in Nature Biotechnology, also introduces a powerful and versatile technology to control the expression of mRNA drugs, which the researchers demonstrate can enhance the effectiveness of mRNA cancer vaccines in preclinical studies of lymphoma. The paper is titled “mRNA vaccine immunity is enhanced by hepatocyte detargeting and not dependent on dendritic cell expression.”
The findings provide a new framework for designing mRNA vaccines and mRNA therapeutics, with immediate implications for cancer immunotherapy, infectious disease vaccines, and gene-editing treatments.

The first human trial of epigenetic reprogramming is underway.
Scientists are testing whether three genes can make old cells behave like young ones again while avoiding the cancer risk that has challenged the field for years.
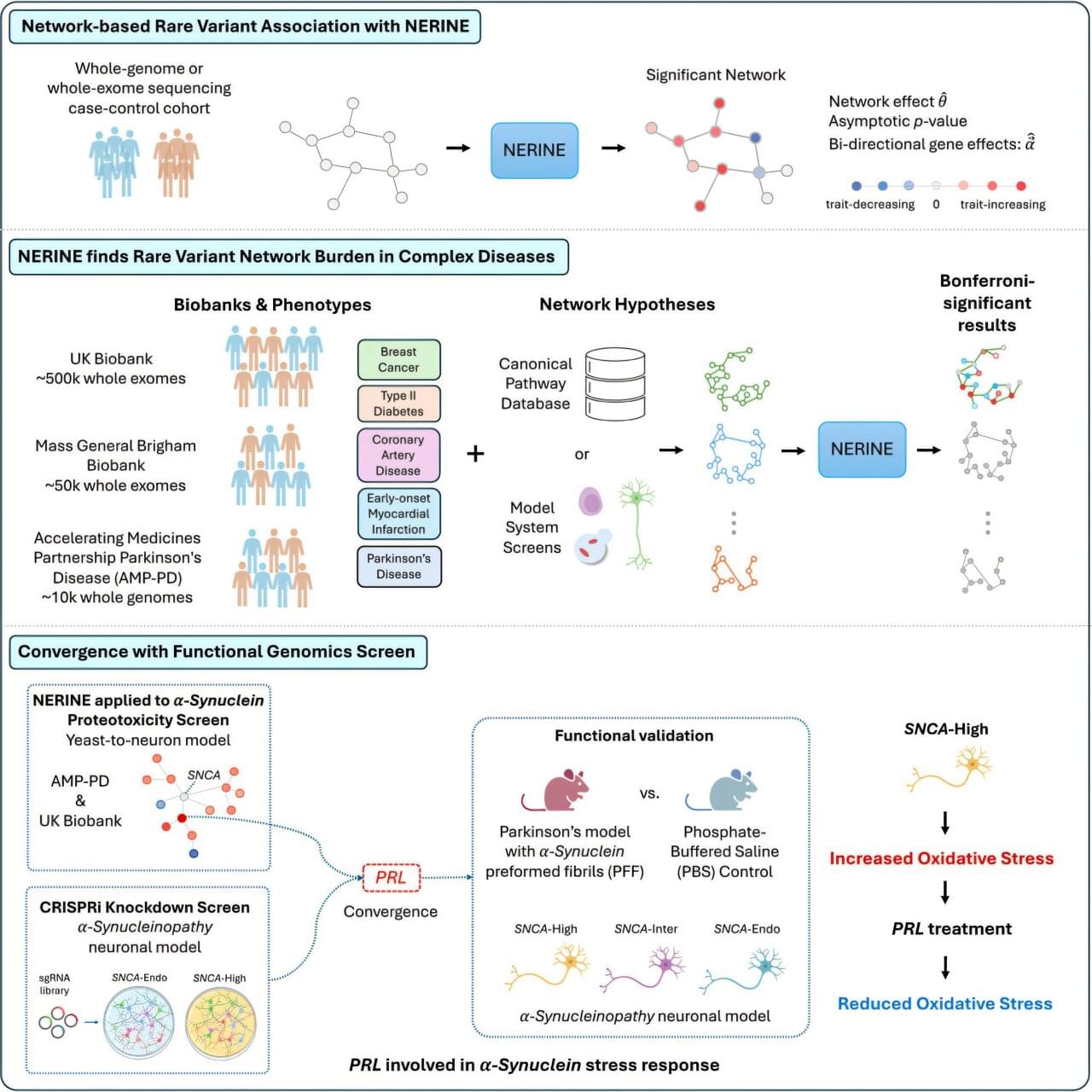
Studies of genetics conducted in yeast cells, human neurons, mice or other model systems often reveal networks of genes that could contribute to complex diseases, such as breast cancer, type 2 diabetes and Parkinson’s disease. But those findings don’t always translate to human biology. Human genetics offers a path to determining which genes among those networks are most relevant to human disease.
Researchers at Harvard Medical School have developed a new statistical framework to link networks identified in models with human genetic data. This could make it faster and easier for researchers to identify which groups of genes are most likely to contribute to a particular human disease, uncover rare disease-causing mutations and zero in on promising therapeutic targets.
The work was published in Cell Genomics.
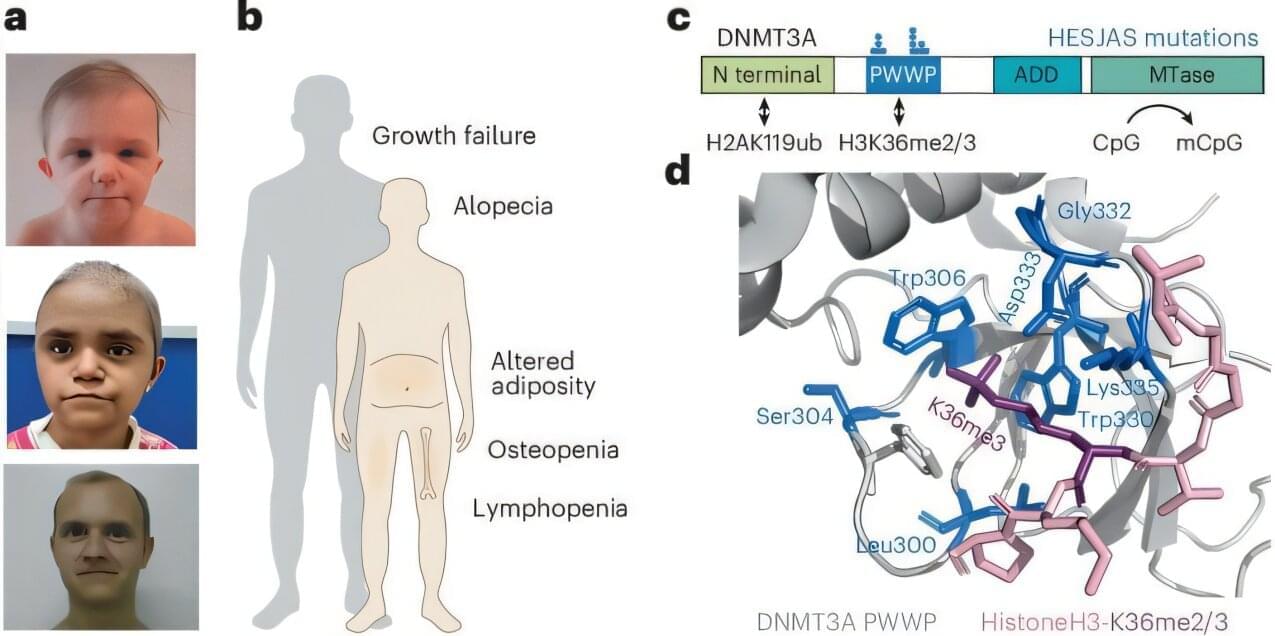
Scientists have discovered a rare genetic condition that causes people to age at a much faster rate, offering fresh insights into the aging process. The study shows for the first time how a “biological clock” present in every cell of the body could contribute to age-related diseases.
Experts say the findings could support the design of future medicines to counter diseases linked to older age, as life expectancies continue to rise across the globe.
The study is published in the journal Nature Genetics.
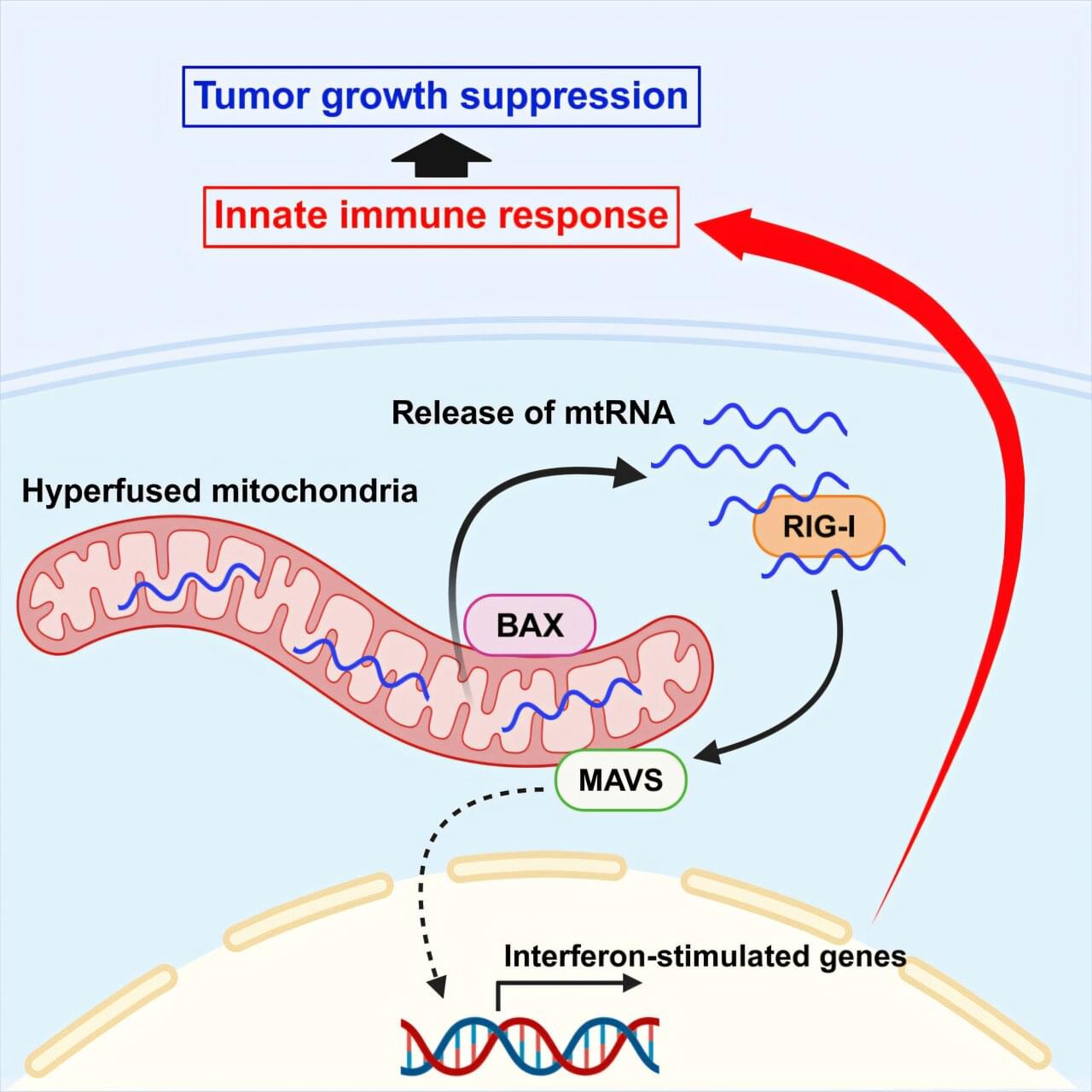
Researchers from the University of Osaka have demonstrated that mitochondrial hyperfusion, when induced by low levels of DRP1 or cellular stress, activates an immune response through the RIG-I–MAVS pathway. Dependent on the involvement of the BAX protein, the release of mitochondrial RNA into the cytosol enhanced natural killer cell cytotoxicity and reduced tumor growth in a xenograft model. The findings, published in Cell Reports, provide new possibilities for cancer research and treatment.
Mitochondria are constantly dividing and fusing within our cells, reshaping themselves to keep up with the cell’s changing needs. Sometimes, though, things go awry, and mitochondria can grow abnormally long. Are these long mitochondria harmful, or might they serve a purpose?
Mitochondria also act as signaling centers, helping the cell sense and respond to trouble. When mitochondria are hyperfused, for example in the stressed, abnormally long state described above, they release their genetic material into the cytosol, where the cell treats it as a warning sign in the same way it would treat a virus.