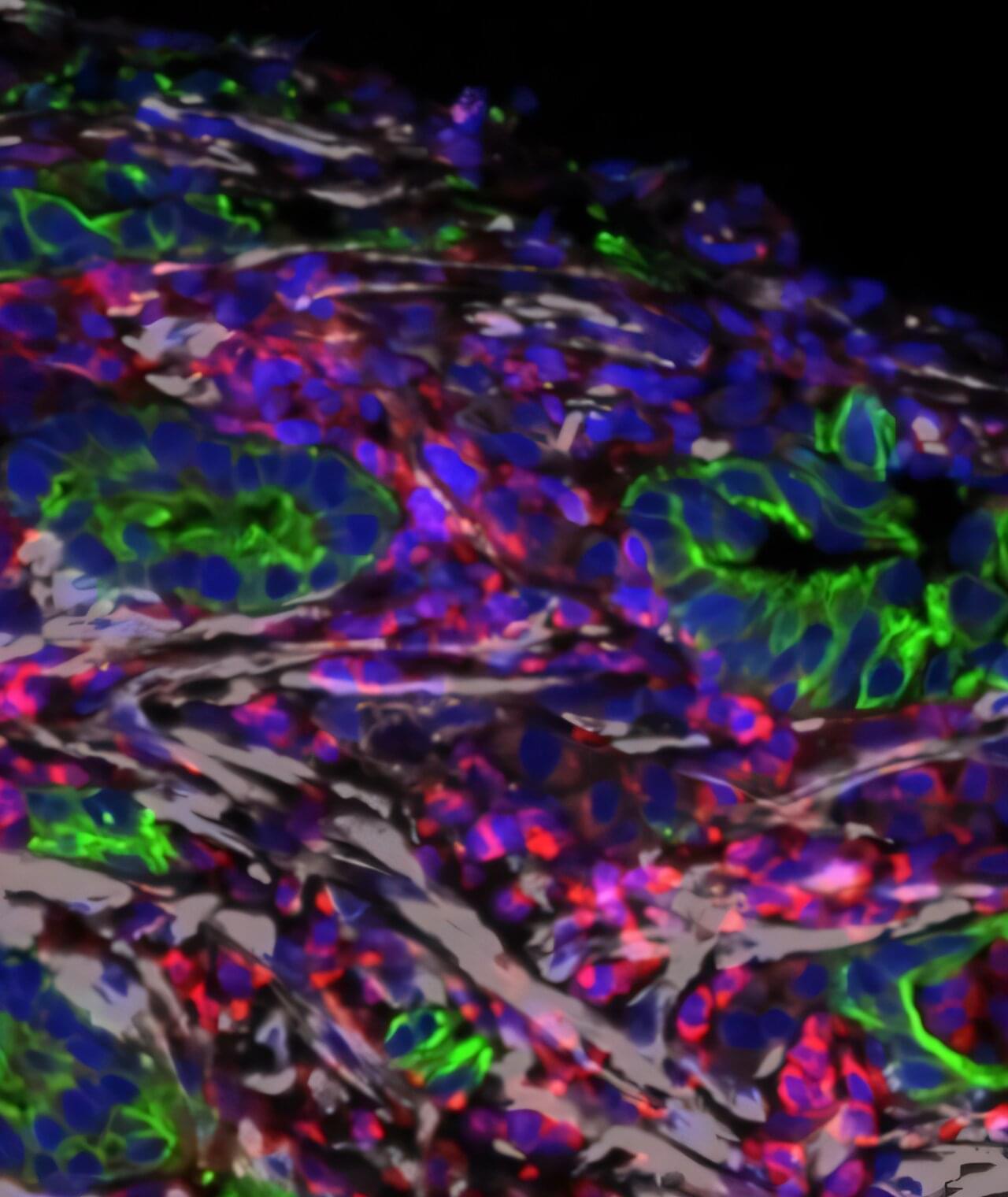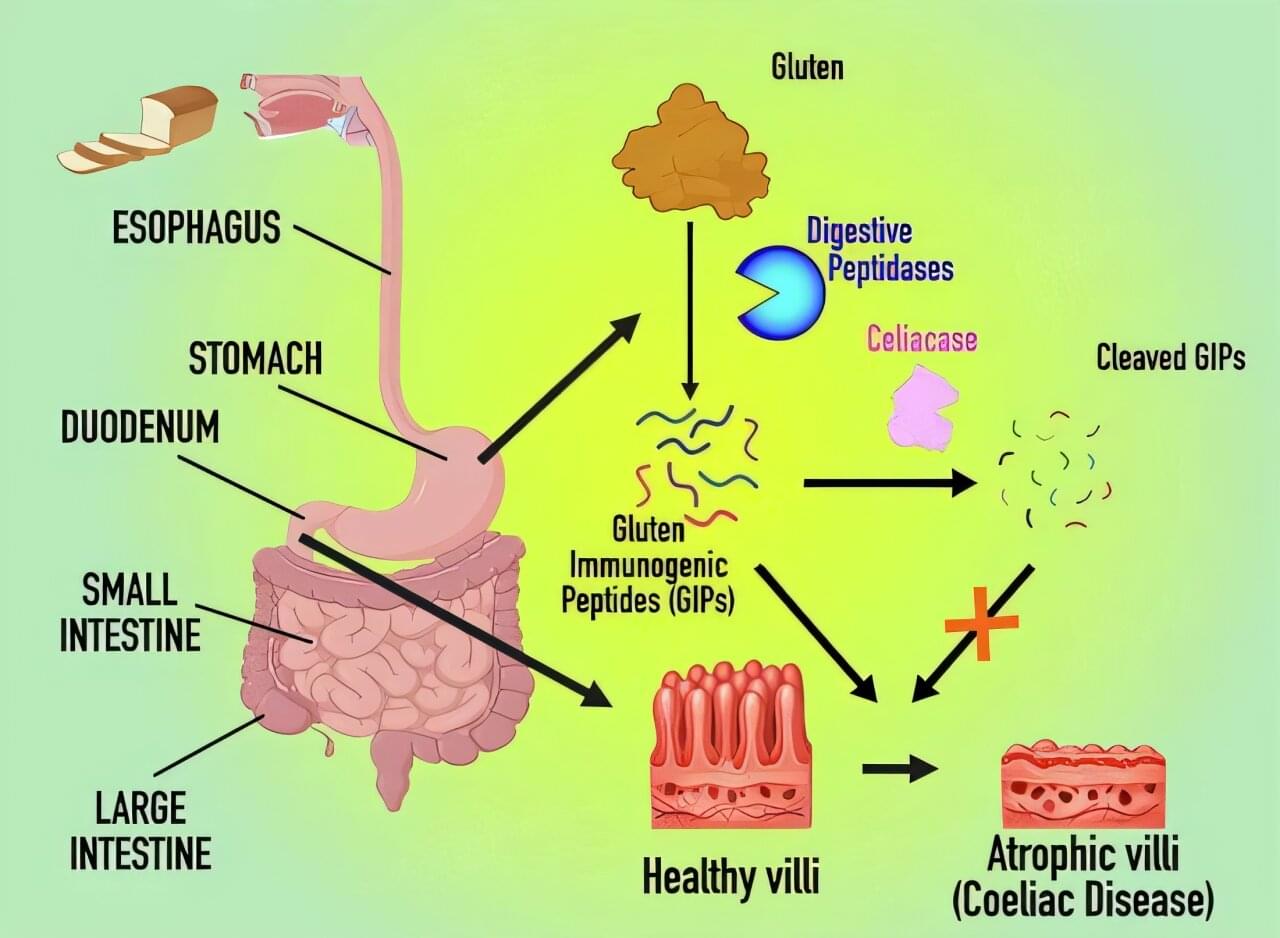
A research project led by the Institute for Research in Nutrition and Food Safety (INSA) and the Faculty of Pharmacy and Food Sciences at the University of Barcelona, together with the Molecular Biology Institute of Barcelona (IBMB) of the CSIC (which stands for Consejo Superior de Investigaciones Científicas), has successfully designed and tested a gluten-degrading molecule that is a promising ally in the management of celiac disease, an autoimmune disease whose symptoms are triggered by the consumption of gluten and other prolamins found in cereals.
At present, there is a complete lack of treatment options beyond a diet free from gluten, which is difficult to maintain in Western societies where diets rely heavily on wheat products.
The major breakthrough is that the molecule is effective at very low concentrations and at a pH of 2—the pH of the stomach—a condition that none of the molecules currently available or under development had previously achieved with efficiency. Although some of them are marketed as nutritional supplements, they are not an effective alternative to gluten-free diets.