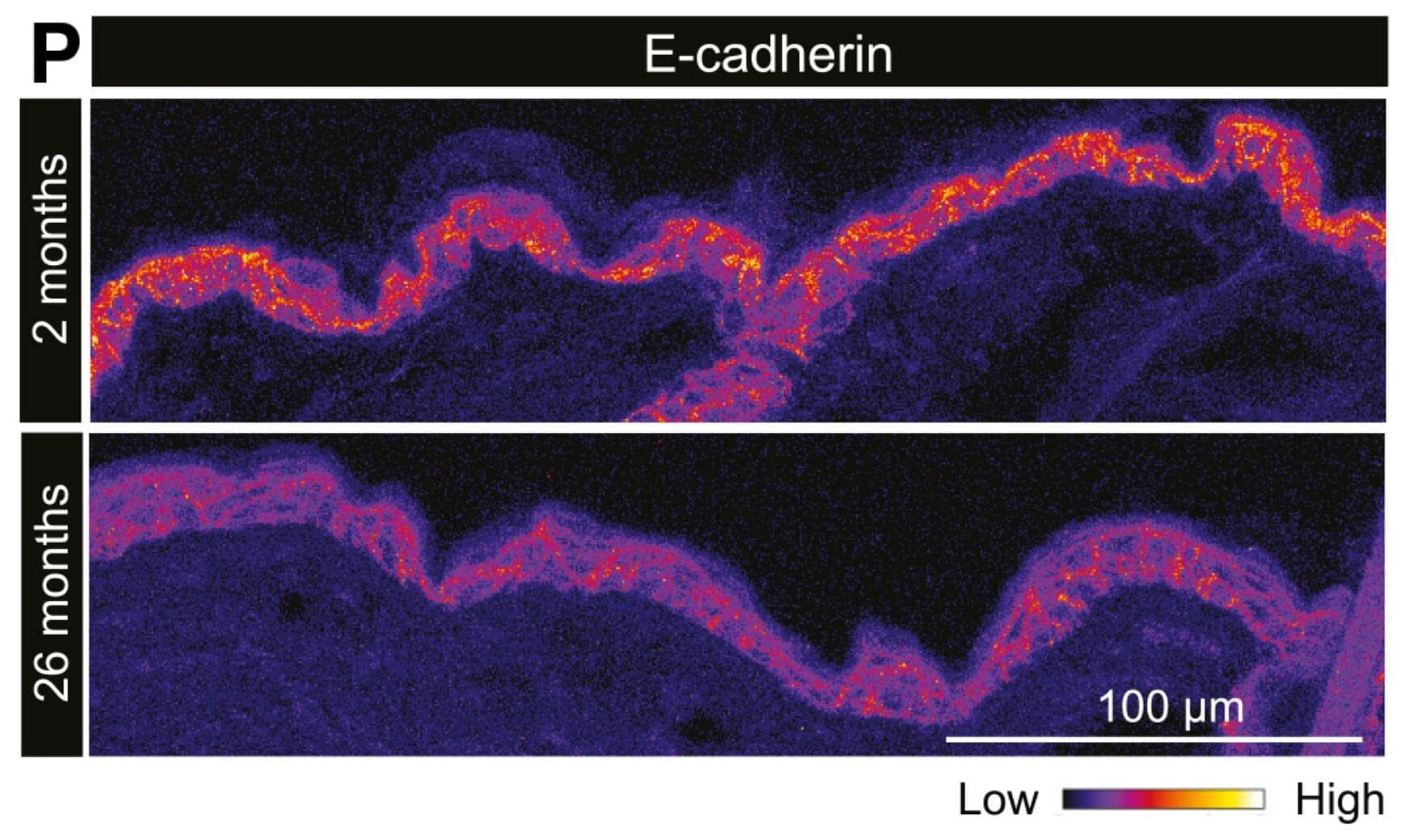
People who carry the APOE2 version of the apolipoprotein E gene are more likely to live to advanced age and are partly protected against Alzheimer’s disease, but scientists have struggled to explain why. A new study from the Buck Institute for Research on Aging, now published in Aging Cell, offers a mechanistic answer: APOE2 helps human neurons keep their DNA intact and resist becoming senescent, a damaged, dysfunctional state that accumulates with age and contributes to neurodegeneration.
The findings shift attention away from APOE’s well-known role in cholesterol transport and toward a previously underappreciated function of the gene: shaping how brain cells maintain the integrity of their genome as they age.
“We’ve known for years that APOE2 carriers tend to live longer and have a lower risk of Alzheimer’s, but the protective mechanism has been a black box,” says senior author Lisa M. Ellerby, Ph.D., professor at the Buck Institute. “Our work shows that APOE2 neurons are better at preventing and repairing DNA damage, and they resist the cellular aging program that drives so much of late-life decline. Our findings point to entirely new therapeutic directions.”