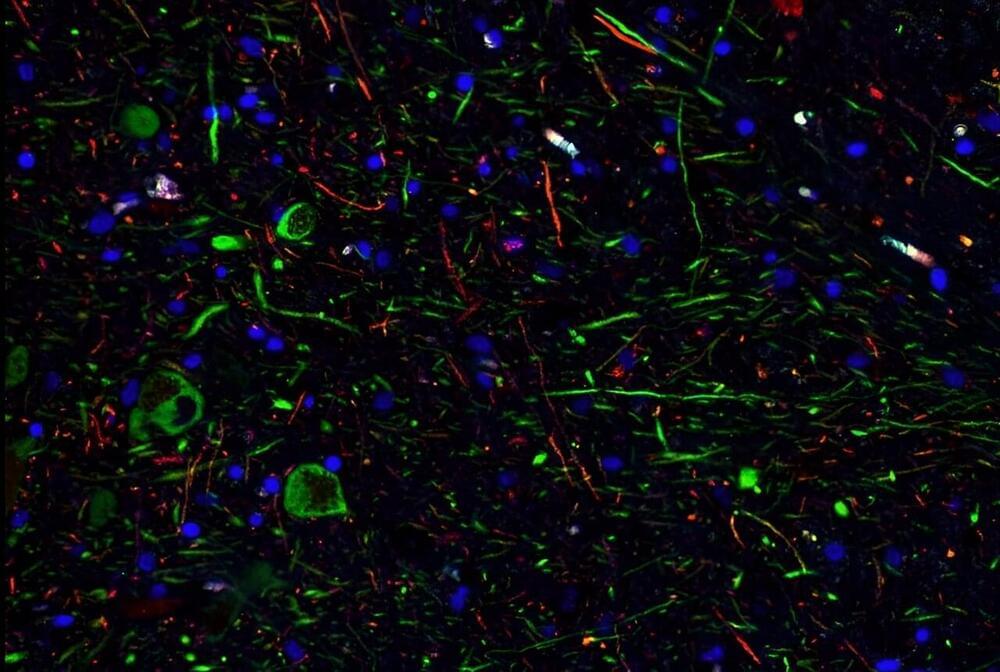
A new study reported that SARS-CoV-2, the virus that causes COVID, can infect dopamine neurons in the brain and trigger senescence—when a cell loses the ability to grow and divide. The researchers from Weill Cornell Medicine, Memorial Sloan Kettering Cancer Center, and Columbia University Vagelos College of Physicians and Surgeons suggest that further research on this finding may shed light on the neurological symptoms associated with long COVID, such as brain fog, lethargy, and depression.
The findings, published in Cell Stem Cell on Jan. 17, show that dopamine neurons infected with SARS-CoV-2 stop working and send out chemical signals that cause inflammation. Normally, these neurons produce dopamine, a neurotransmitter that plays a role in feelings of pleasure, motivation, memory, sleep, and movement. Damage to these neurons is also connected to Parkinson’s disease.
“This project started out to investigate how various types of cells in different organs respond to SARS-CoV-2 infection. We tested lung cells, heart cells, pancreatic beta cells, but the senescence pathway is only activated in dopamine neurons,” said senior author Dr. Shuibing Chen, director of the Center for Genomic Health, the Kilts Family Professor Surgery and a member of the Hartman Institute for Therapeutic Organ Regeneration at Weill Cornell Medicine. “This was a completely unexpected result.”







{kind=link}