Who cares about running DOOM on a pregnancy test?
Category: neuroscience – Page 100
Enhancing gut-brain communication reversed cognitive decline, improved memory formation in aging mice
The sight of a delectable plate of lasagna or the aroma of a holiday ham are sure to get hungry bellies rumbling in anticipation of a feast to come. But although we’ve all experienced the sensation of “eating” with our eyes and noses before food meets mouth, much less is known about the information superhighway, known as the vagus nerve, that sends signals in the opposite direction — from your gut straight to your brain.
These signals relay more than just what you’ve eaten and when you are full. A new study in mice from researchers at Stanford Medicine and the Palo Alto, California-based Arc Institute has identified a critical link between the bacteria that live in your gut and the cognitive decline that often occurs with aging.
“Although memory loss is common with age, it affects people differently and at different ages,” said Christoph Thaiss, PhD, assistant professor of pathology. “We wanted to understand why some very old people remain cognitively sharp while other people see significant declines beginning in their 50s or 60s. What we learned is that the timeline of memory decline is not hardwired; it’s actively modulated in the body, and the gastrointestinal tract is a critical regulator of this process.”
Aging causes changes in gut bacteria in mice, which hampers communication between the intestines and the brain. Restoring this connection helped old mice form memories as well as young animals.
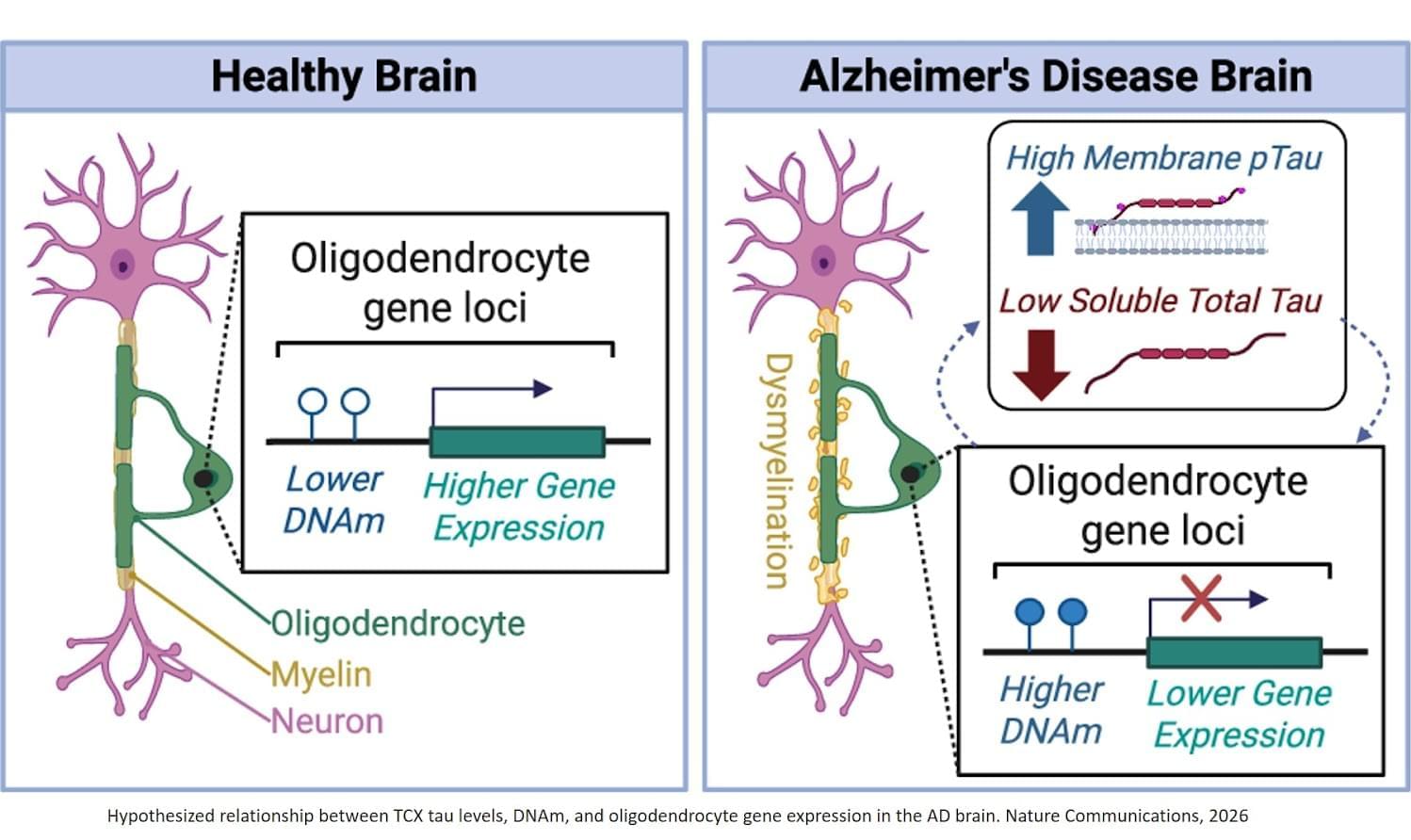
Oligodendrocyte molecular perturbations associated with tau in Alzheimer’s
The findings suggest that in AD, part of what happens in the brain may involve changes in DNA tagging that affect the function of oligodendrocytes, particularly in relation to the buildup of the toxic protein tau.
Oligodendrocytes are the brain cells that make myelin, the insulation that helps nerve cells communicate. Scientists have theorized that disrupting neuron communication contributes to symptoms for people with AD. Researchers in this study found that nearly all significant methylation changes — small chemical tags added to DNA that help control when genes are turned on or off — were linked to the tau protein. This supports the idea that this protein plays a key role in brain cell changes tied to AD.
“Our team has previously shown that oligodendrocytes are affected in Alzheimer’s and another tau-related disease, progressive supranuclear palsy (PSP),” says the author. “These new results further highlight that problems in oligodendrocytes and myelin are central to AD. They also point to specific molecular pathways, particularly epigenetic changes, that could be targeted in future therapies.”
The study results identified new genes that may play a role in AD, including one called LDB3, and confirmed many findings across multiple independent datasets, showing its reliability. The identification of specific genes provides potential targets for future research — for example, scientists might investigate whether interventions that reverse methylation or support oligodendrocyte health can slow or modify disease progression for patients with AD. ScienceMission sciencenewshighlights.
In a study published in Nature Communications, the researchers have identified specific DNA-level changes in the brains of people with Alzheimer’s disease (AD). Using advanced biological analysis, the team mapped alterations in the brain’s regulatory landscape that may help explain why Alzheimer’s presents and progresses differently from person to person. The findings could also open new avenues for understanding other neurodegenerative diseases.
Alzheimer’s disease is the most common cause of dementia. Biologically, the disease begins with the formation of protein deposits, known as amyloid plaques, and neurofibrillary tangles in the brain. This causes brain cells to die over time and the brain to shrink. About 6.9 million people in the U.S. age 65 and older live with Alzheimer’s disease. There is no cure, and in advanced stages, complications can result in a significant decline in quality of life and death.
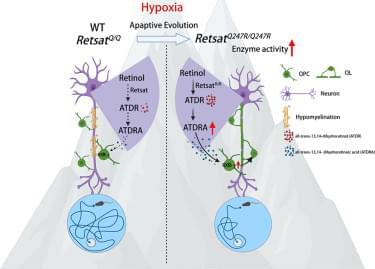
A gain-of-function Retsat variant from high-altitude adaptation promotes myelination via a neuronal dihydroretinoic acid-RXR-γ pathway
High-altitude survival gene in mammals may help reverse nerve damage from conditions like multiple sclerosis.
Neuron.
Li et al. report that a gain-of-function Retsat variant, associated with high-altitude adaptation, promotes myelination by boosting neuronal synthesis of the signaling metabolite ATDRA. This molecule activates RXR-γ in oligodendrocyte progenitors. Administration of the prodrug ATDR promotes remyelination in models of myelin disease.

How the brain can selectively focus attention on one voice among others in a noisy environment
MIT neuroscientists have figured out how the brain is able to focus on a single voice among a cacophony of many voices, shedding light on a longstanding neuroscientific phenomenon known as the “cocktail party problem.”
This attentional focus becomes necessary when you’re in any crowded environment, such as a cocktail party, with many conversations going on at once. Somehow, your brain is able to follow the voice of the person you’re talking to, despite all the other voices that you’re hearing in the background.
Using a computational model of the auditory system, the MIT team found that amplifying the activity of the neural processing units that respond to features of a target voice, such as its pitch, allows that voice to be boosted to the forefront of attention.

Stress rewires brain control networks, boosting pain tolerance in ice test
Stress resilience isn’t a flatline. It’s a flex, according to new research from Florida International University. Marcelo Bigliassi, assistant professor of psychophysiology, and Ph.D. student Dayanne Antonio thrive in creating stressful environments.
They set out to explore how the brain’s internal wiring and a person’s subjective experience of stress interact to determine how they respond to stressful situations. The findings were published in the Journal of Applied Physiology.
Study participants plunged one hand into a bucket of ice-cold water. Frigidly cold water. As the seconds ticked by, the body’s stress systems revved up, and their skin began to sweat.