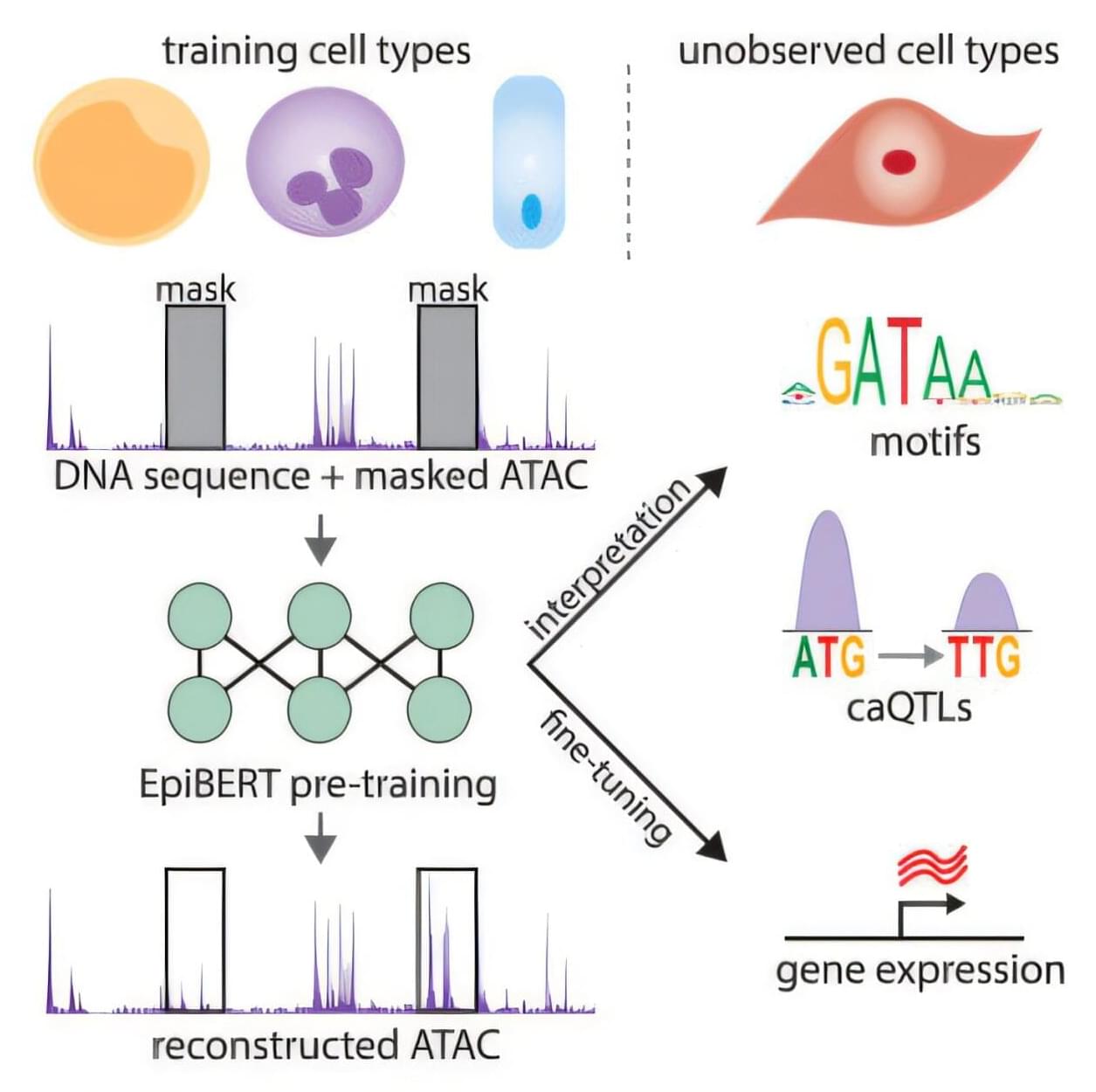
In today’s AI news, Block announced the launch of Goose, an open-source AI agent that allows developers to customize the tool for different purposes and using different large language models. Block’s move comes just after the Chinese startup DeepSeek unveiled its R1 artificial intelligence model, a rival to leading U.S. AI providers.
In other advancements, Cerebras and the Mayo Clinic, announcing a joint project for AI tools used in patient care. The technology was announced at the JP Morgan Healthcare Conference. In this project, the stakeholders aim to use a human reference genome to combine with patient data in order to try to identify genetic differences.
S innovative R1 reasoning model into its platform to revolutionize AI-powered search. This strategic integration strengthens Perplexity’s ability to perform deep web searches, providing users with more comprehensive and accurate results while upholding strict data security standards. + And, San Francisco-based start-up Atomicwork will today announce it has clinched $25 million of Series A financing in a round led by Khosla Ventures and Z47. The funding announcement comes less than six months after the last round, takes the total amount of money raised by the company since its launch to almost $40 million.
In videos, on this episode of Top of Mind, Gartner Global Chief of Research Chris Howard is joined by Rita Sallam, Distinguished VP Analyst, to explore how organizations can create measurable value with AI in 2025. Learn how to align investments with business priorities, manage productivity challenges and balance risk with ambition.
Ll gain a deeper knowledge of what makes a chatbot truly effective. + And, AI is transforming science in ways we never imagined. Bonnie Kruft, Deputy Director of AI for Science at Microsoft Research, shares how cutting-edge AI models are revolutionizing drug discovery, materials science, and climate prediction. Bonnie reveals how these innovations are shaping a healthier, more sustainable future for all.
S open-source model allows sharing of information, helping to lift all AI progress. He joins Caroline Hyde and Mike Shepard on “Bloomberg Technology” to discuss. ‘ + Thats all for today, but AI is moving fast — like, comment, and subscribe for more AI news! Please vote for me in the Entrepreneur of Impact Competition today! Thank you for supporting my partners and I, it’s how I keep Neural News Network free.
[](https://open.substack.com/pub/remunerationlabs/p/block-launc…hare=true)