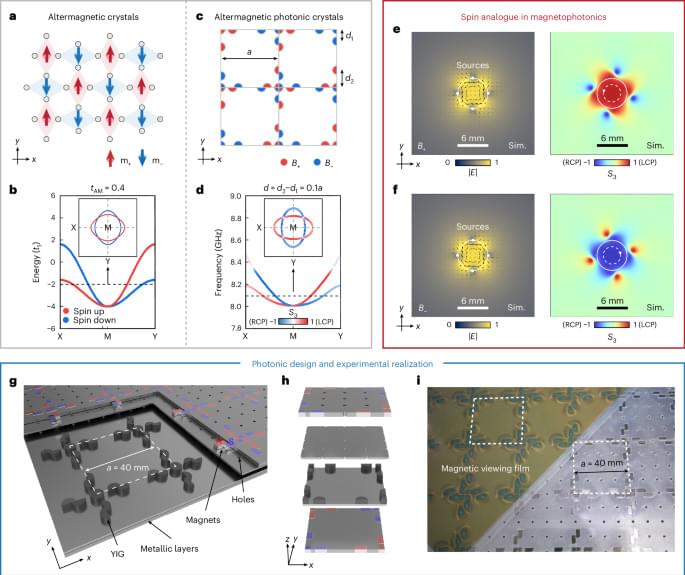
By training an AI model using 153,208 plasmids from Addgene, Shao et al.’s PlasmidGPT can annotate and classify existing plasmids as well as generate new functional plasmid sequences from DNA “prompts”. While existing plasmid design tools currently surpass PlasmidGPT in sophistication, future architecture and training data augmentations may allow us to automate much of the plasmid design process. I could certainly see this playing a role in high-throughput biological screening methods.
Assembly standards facilitate the construction of functional plasmids (13). Collections such SEVA (14, 15) and CIDAR MoClo (16) include ready-to-use constructs and genetic parts that can be easily assembled, making them valuable tools for microorganism bioengineering and genome editing. Tools such as Cello (17, 18) can design genetic constructs with a high success rate for specific functions, such as computation, across diverse organisms. iBioSim 3 enables the design and modeling of genetic circuits that extend beyond logic circuits (19). However, there is still no computational method capable of harnessing the existing collection of plasmid sequences for designing the full spectrum of plasmids, such as those for mammalian expression, bacterial expression, and gateway vectors. Consequently, for many applications, plasmid DNA design remains a labor-intense process that requires manual inspection, annotation, and the combination of functional sequences.
Recently, generative models such generative pretrained transformers (GPTs) (20) have demonstrated remarkable success in modeling human language. Given the similarity of human language and biological sequences such as protein and DNA, researchers have adapted these frameworks to design proteins (21) and, more recently, to generate genomic sequences that contain potentially functional regulatory elements and genes (22–24). Despite these advances, it remains an open question whether language models can be leveraged to efficiently design and analyze complex engineered DNA.
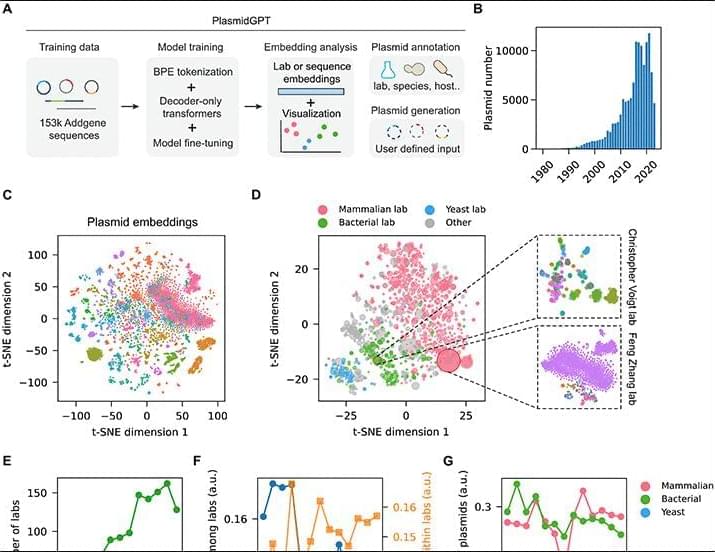
Here, we introduce PlasmidGPT, a generative framework for designing and annotating plasmid DNA sequences (Fig. 1A). Our framework is built on a decoder-only transformer model that is pretrained on 153,208 plasmid sequences from Addgene (25), a public repository for engineered DNA sequences. We demonstrate that sequence embeddings generated by PlasmidGPT encode plasmid sequences into a continuous numerical space. These sequence representations facilitate the visualization of research topics across laboratories by capturing sequence-level similarities and variations. Leveraging simple machine learning models trained on these embeddings, PlasmidGPT enables the fast identification of a wide range of high-level plasmid features (vector type, selectable marker, growth strain, and lab of origin) directly from sequence, facilitating plasmid analysis tasks such as functional annotation and provenance tracking. Moreover, PlasmidGPT generates plasmids that have genetic part distributions similar to those of the training sequences. Conditional plasmid generation can be achieved either by providing a user-specified starting sequence or by fine-tuning the model using special tokens that represent specific vector types. Furthermore, we experimentally validated the functionality of two model-generated plasmids in bacterial cells.