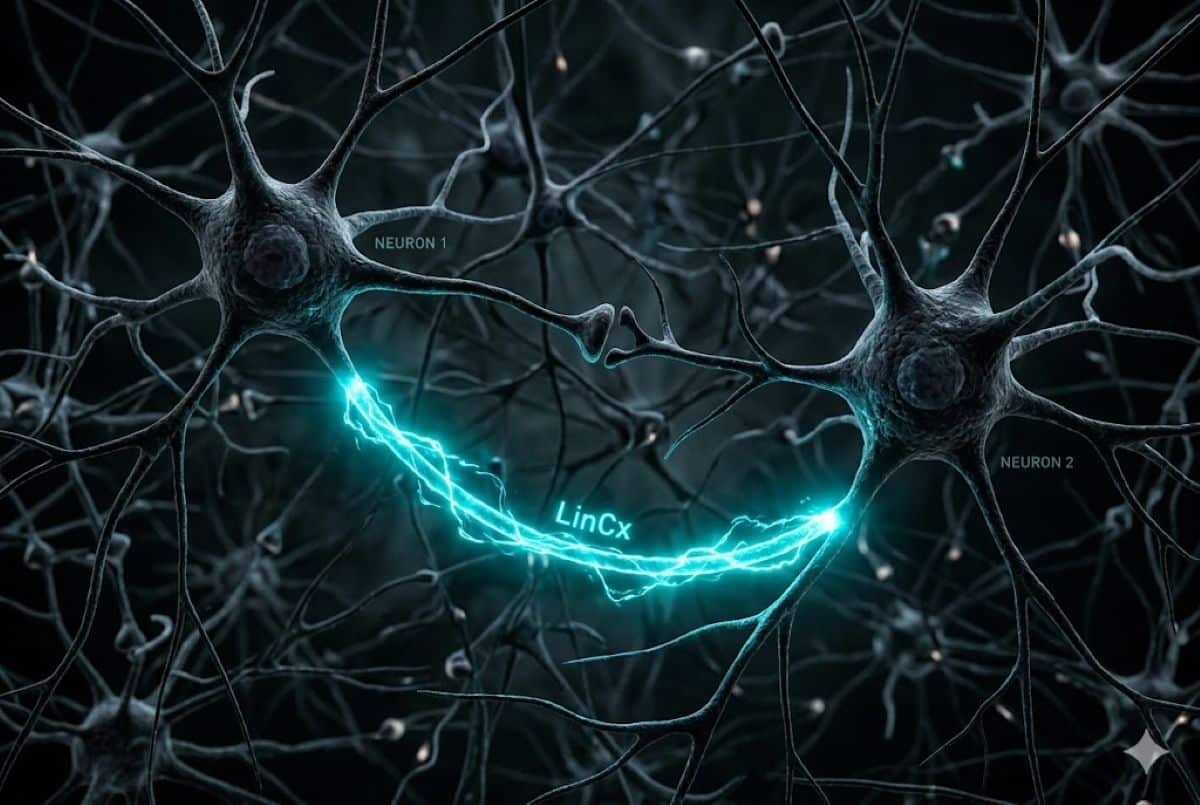
Researchers develop LinCx, a biological “wire” that creates precision electrical bypasses in the brain to restore function without drugs or external stimulation.

The relationship between sleep and disease suggests that there exists a connection between the brain and the body that extends beyond merely influencing the brain itself.
Among brain-related disorders, short sleep was significantly associated with depressive episodes and anxiety disorders, as seen in other studies of sleep and mental health. Short sleep was also associated with obesity, type 2 diabetes, hypertension, ischemic heart disease, and heart arrhythmias.
Short and long sleep were associated with chronic obstructive pulmonary disease, asthma, and a cluster of digestive disorders, including gastritis and gastroesophageal reflux disease.

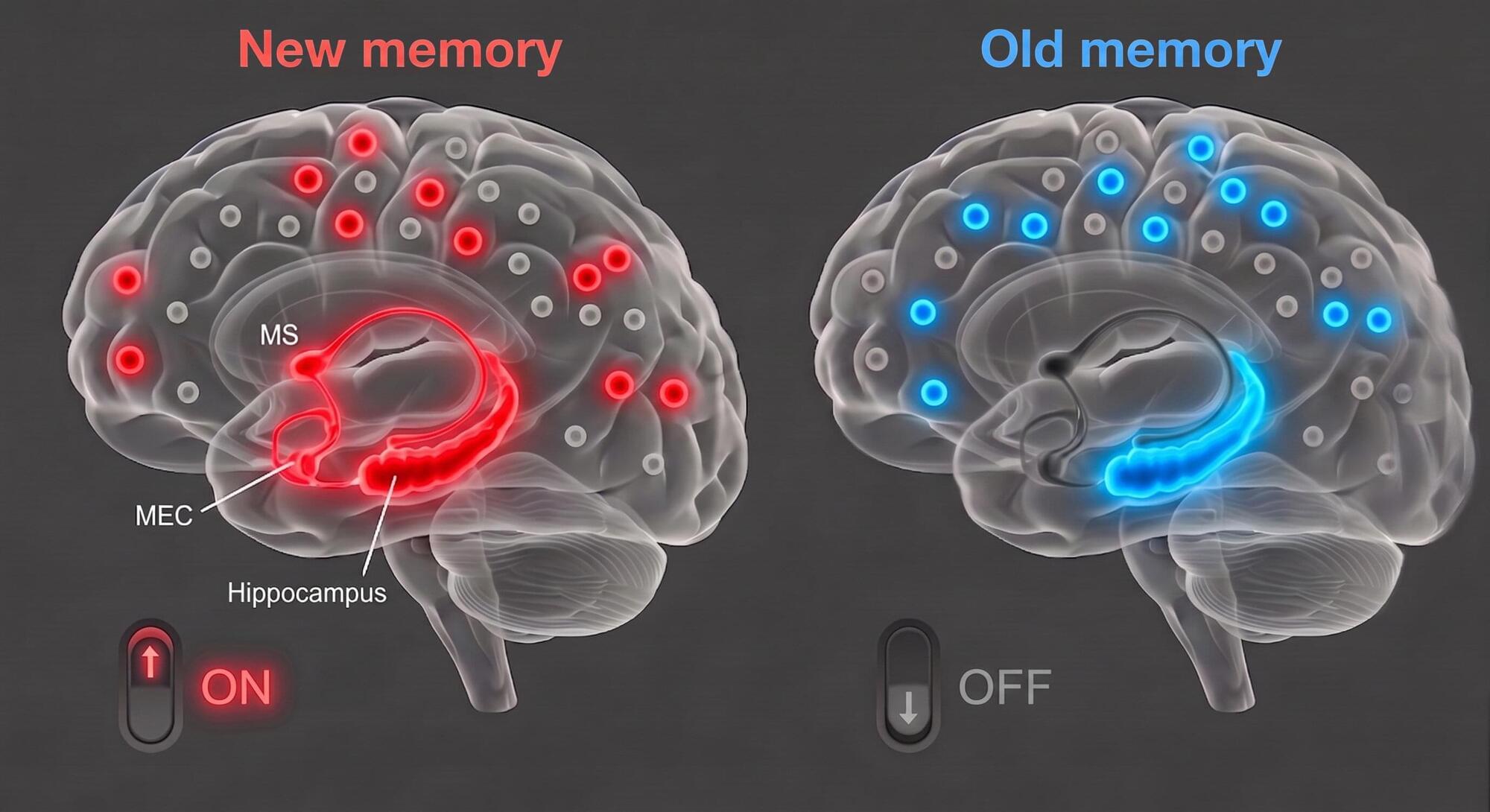
As humans and other animals experience new things, their brains continuously update their memory of past events. These updates allow them to adapt to changing environments, all while preserving older memories that could still help them to make decisions in some situations.
Many past neuroscience studies have investigated the neural circuits involved in the encoding and retrieval of memories. However, the mechanisms via which it decides whether to retrieve older or newly updated memories remain poorly understood.
Researchers at Korea Advanced Institute of Science and Technology (KAIST) recently carried out a study involving mice that was aimed at better understanding how the brain switches between older and newer memories.
Sponsors: Accelerate your efficiency. Sign up for your one-dollar-per-month trial today at http://shopify.com/theories Sign up for Claude today at http://claude.ai/theoriesofeverything and checkout Claude Pro — which includes access to all of the features mentioned in today’s episode. I personally subscribe to The Economist. FLASH SALE to May 18: 50% off annual (double the usual!) No other podcast has this! https://economist.com/TOE
What if gravity is just entropy in disguise? Professor Erik Verlinde joins me to argue that gravity isn’t a fundamental force—it’s thermodynamic, emerging from quantum information the way gas pressure emerges from molecules bouncing around. We explore why spacetime may be stitched together by entanglement, and how dark energy and dark matter both pop out automatically without extra particles or parameters. Verlinde explains why the cosmological constant problem is a red herring, and why there may be no final theory of physics. When asked where the universe comes from, his answer is one word: chaos.
SUPPORT: Support me on Substack: https://curtjaimungal.substack.com/su… me on Crypto: https://commerce.coinbase.com/checkou… Support me on PayPal: https://www.paypal.com/donate?hosted_… JOIN MY SUBSTACK (Personal Writings): https://curtjaimungal.substack.com LISTEN ON SPOTIFY: https://open.spotify.com/show/4gL14b9… TIMESTAMPS:
LINKS MENTIONED: Papers, books, websites:
Videos:
SOCIALS:
Guests do not pay to appear. Theories of Everything receives revenue solely from viewer donations, platform ads, and clearly labelled sponsors; no guest or associated entity has ever given compensation, directly or through intermediaries. #science.
JOIN MY SUBSTACK (Personal Writings): https://curtjaimungal.substack.com.
LISTEN ON SPOTIFY: https://open.spotify.com/show/4gL14b9…
TIMESTAMPS: 00:00:00 — Thermodynamic Gravity and Information 00:06:35 — Beyond Effective Field Theory 00:13:08 — Turtles All The Way Down 00:25:41 — Entropy as a Force 00:36:31 — Entanglement and Spatial Connectivity 00:47:31 — Deriving Inertia and F=ma 00:56:41 — De Sitter Space Challenges 01:02:01 — Dark Matter and Milgram 01:11:51 — The Emergence of Time 01:21:01 — Statistical Gravity Fluctuations 01:27:01 — Quantum Computational Complexity 01:36:01 — Physics Intuition and Mentorship 01:47:31 — Beauty, Garbage, and Chaos.