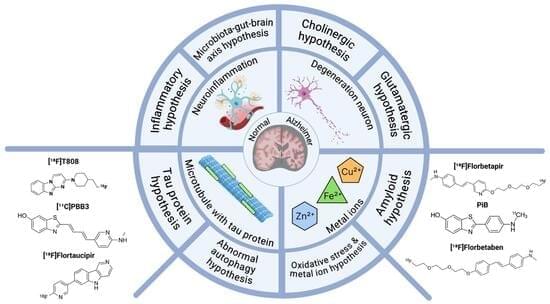
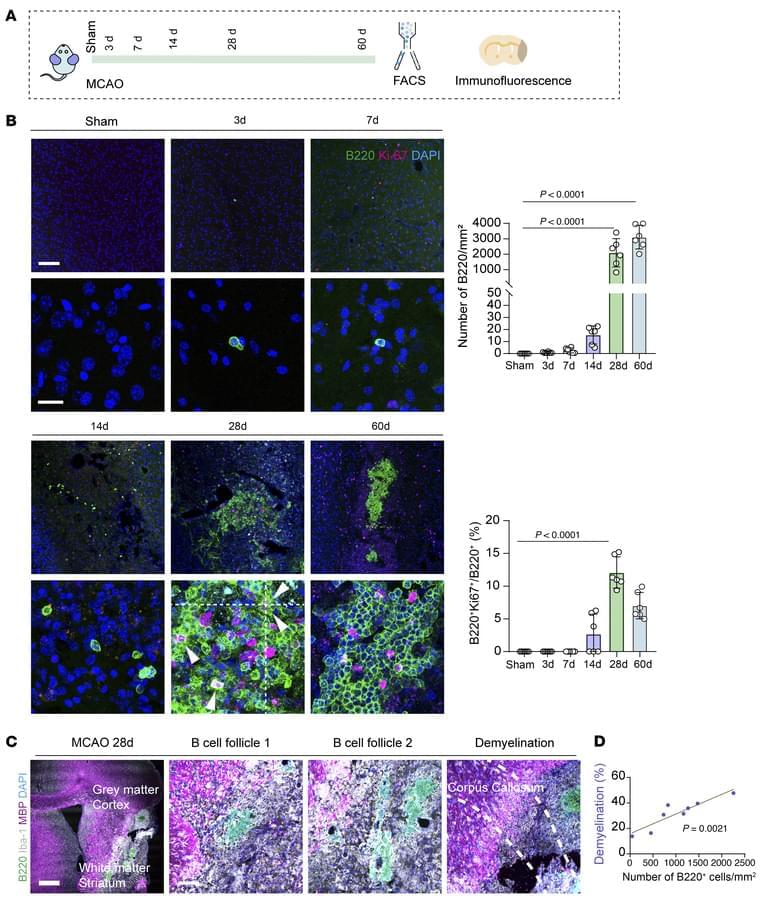
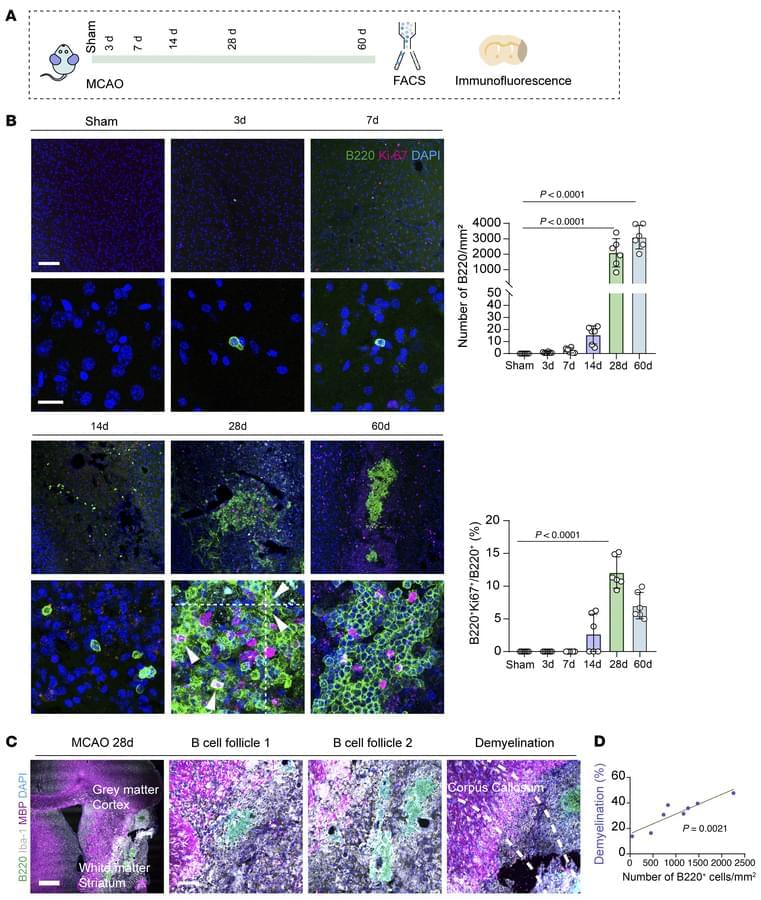
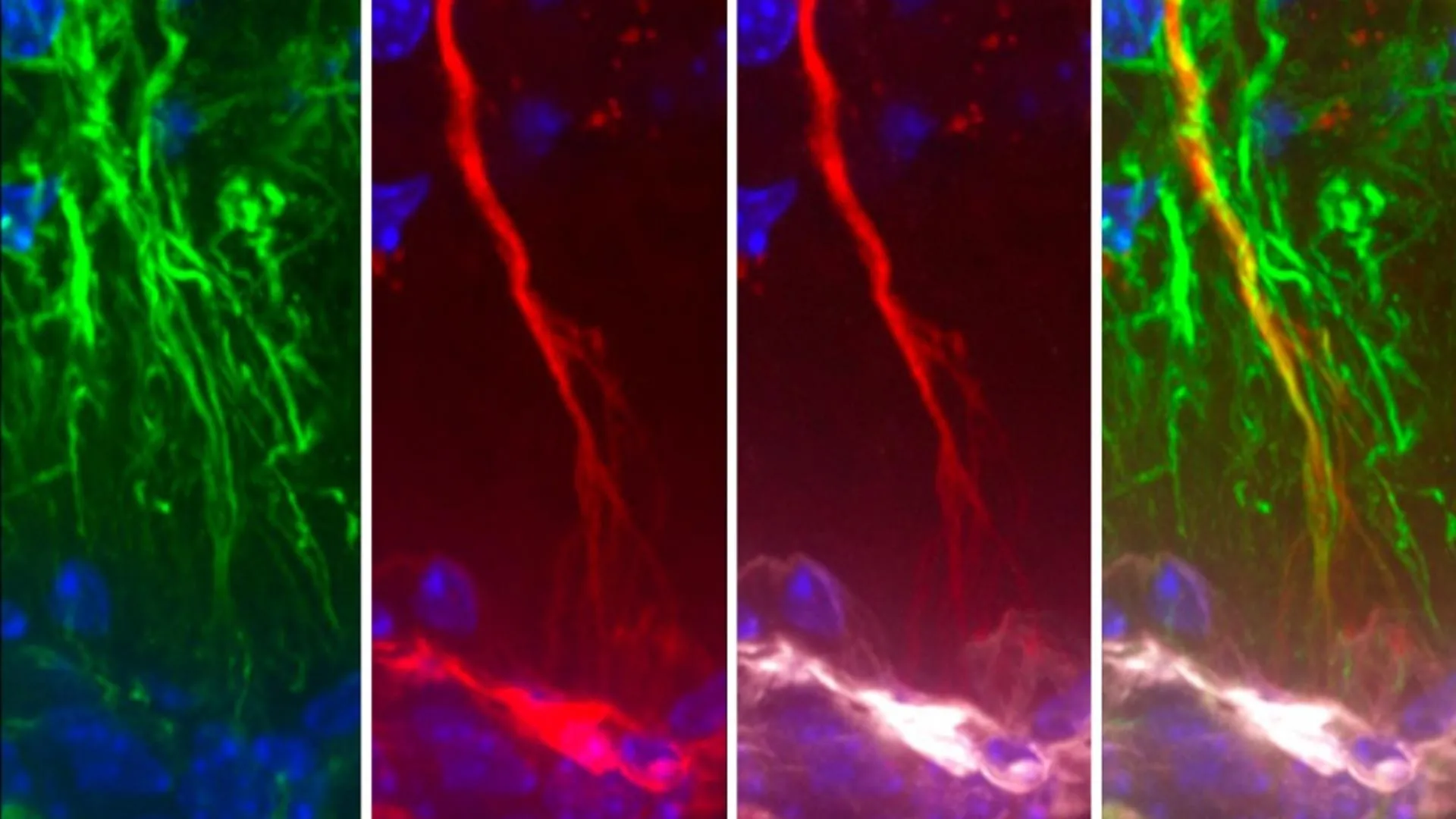
Triggering receptor expressed on myeloid cells 2 (TREM2) is a cell surface transmembrane receptor from the TREM receptor family, predominantly expressed on the microglia in the central nervous system (CNS). TREM2-initiated signaling plays a crucial role in regulating neuroinflammation and neurodegeneration, particularly in the context of neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD), through the activation of downstream signaling pathways and transcriptional regulation of relevant genes. In this review, we aim to provide a concise review of the role and mechanistic implications of TREM2 in neurodegeneration and neuroinflammation, with a specific focus on AD and PD. We will discuss the most recent preclinical studies to highlight current advancements in the field. This review is intended to support both basic researchers and clinicians by enhancing their understanding of microglial function in the pathophysiology of AD and PD, as well as its role in neuroinflammation and neurodegeneration. Ultimately, we hope this contribution will pave the way for new discoveries and the development of potential therapeutic interventions.
© 2026. The Author(s).