Circa 1999 this can lead to genetic editing that allows people to handle even a nuclear fallout level of radiation and even allow them to handle outer space better.
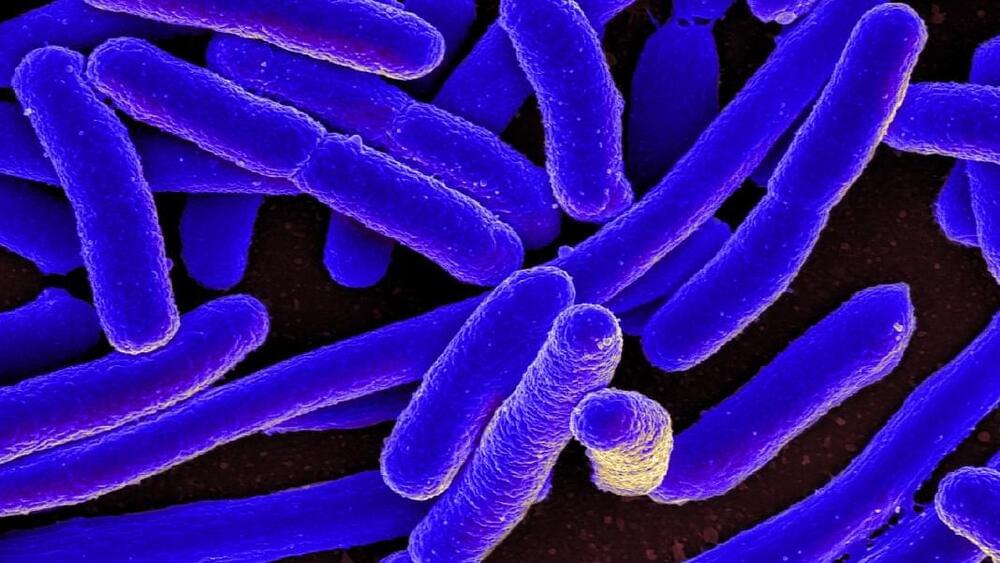
Rockville, MD — No, it’s not the cockroach, but rather a strain of pink bacteria that can survive 1.5 million rads of gamma irradiation — a dose 3,000 times the amount that would kill a human. This dose of radiation shreds the bacteria’s genome into hundreds of pieces. The organism’s remarkable ability to repair this DNA damage completely in a day and go on living offers researchers tantalizing clues to better understanding the mechanism of cellular repair. Advances in this area could in turn improve our understanding of cancer which is frequently caused by unrepaired DNA damage. Genetically engineering the microbe could lead to improved ways to cleanup pollution and to new industrial processes.
U.S. Department of Energy-funded researchers at The Institute for Genomic Research (TIGR) describe the complete genetic sequence of the bacteria Deinococcus radiodurans in the November 19 issue of Science.
“This is a significant accomplishment,” Secretary of Energy Bill Richardson said. “The Department of Energy began microbial genome work to support bold science and to help meet our unique environment and energy mission needs. Besides the insights into the way cells work, this new research may help provide a new safe and inexpensive tool for some of the nation’s most difficult cleanup challenges.”



{kind=link}
{kind=link}