Despite ample evidence for functional specialization in the brains of humans (Kanwisher, 2010) and nonhuman animals (Tsao et al., 2006), some continue to argue against the idea of stable structure in the brain, emphasizing the distributed, dynamic, and interactive nature of cognitive processes, including language (Pessoa, 2022; Forkel and Hagoort, 2024; Drijvers et al., 2025). Deep engagement with this debate is beyond the scope of this article, but two points are worth clarifying. First, the fact that many areas—sometimes in distant parts of the brain—are engaged by language comprehension does not imply that the “entire brain” supports this function. Although the extended language network spans almost every major component of the brain, within each component, language regions occupy a small fraction of brain tissue. Second, linguistic inputs can unquestionably engage many brain regions beyond those that specifically support language processing: vivid descriptions of faces or scenes can engage category-selective visual areas, a story about a misunderstanding can engage the Theory of Mind network, and a horror story can engage the amygdala (see Casto, et al., 2025b for discussion). However, all these brain regions can also be engaged by nonlinguistic inputs. The ability of a brain region to be engaged by language does not make it a “language region” any more than its ability to be engaged by visual inputs makes it a “visual region.” Furthermore, the fact that the language network needs to interact with other brain areas does not undermine its functional distinctness from those areas and its special role in language processing. As long as different components within the language network interact more strongly with one another than with other networks—for which ample evidence exists (Blank et al., 2014; Braga et al., 2020; Du et al., 2024, 2025; Shain and Fedorenko, 2025)—the language network and other cognitive networks can be treated as meaningfully distinct objects of study (Simon, 1962).
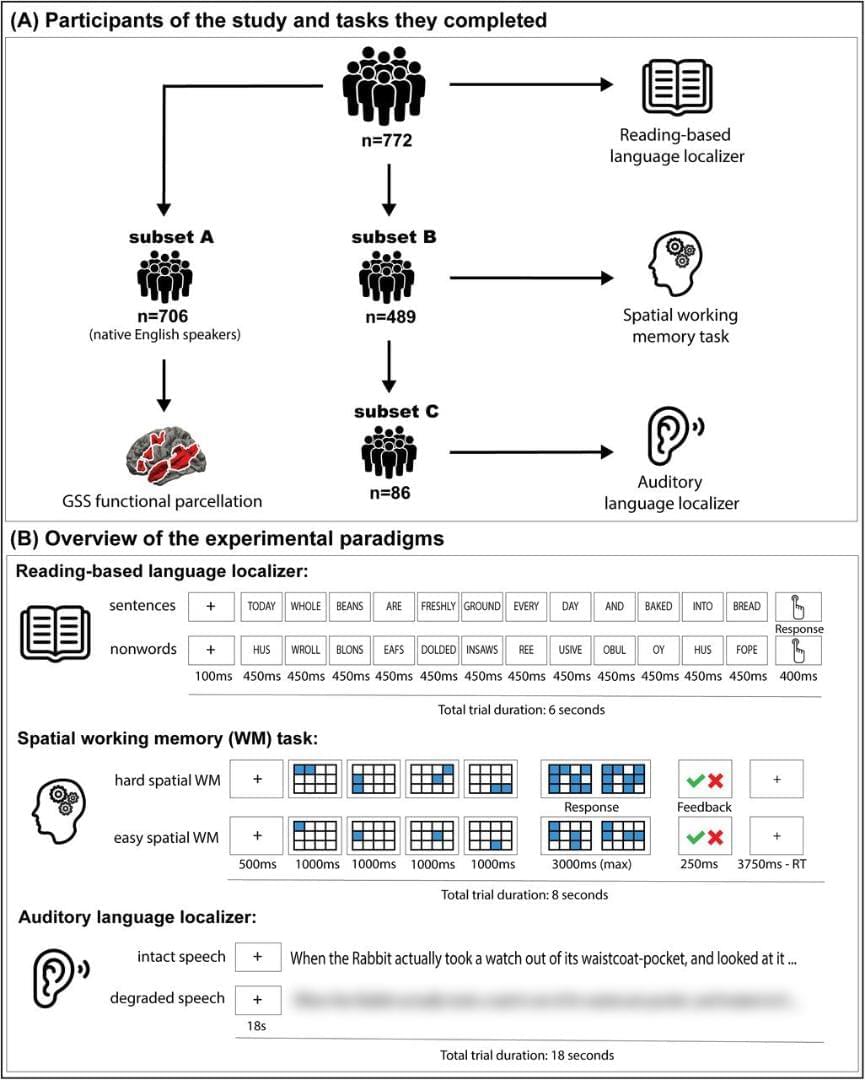
For completeness and ease of comparison with past studies, we explored the possibility of using standard anatomical atlases to constrain individual fROIs (rather than the parcels derived from a group-level representation of brain activity; Fedorenko et al., 2010; Julian et al., 2012). Examining individual activation maps (or maps derived from functional connectivity patterns: Braga et al., 2020; Du et al., 2024, 2025; Shain and Fedorenko, 2025) against standardized brain parcellations reveals two issues. First, individual topographies often do not align with the boundaries of the atlas areas: a contiguous functional region may get broken up by a boundary or—for finer-grained atlases—may get assigned to different atlas areas across individuals because of interindividual topographic variability (see Fig. S5B for examples). For studies focusing on a particular functional network, we therefore recommend functional parcels over anatomical/multimodal atlases.


{kind=link}