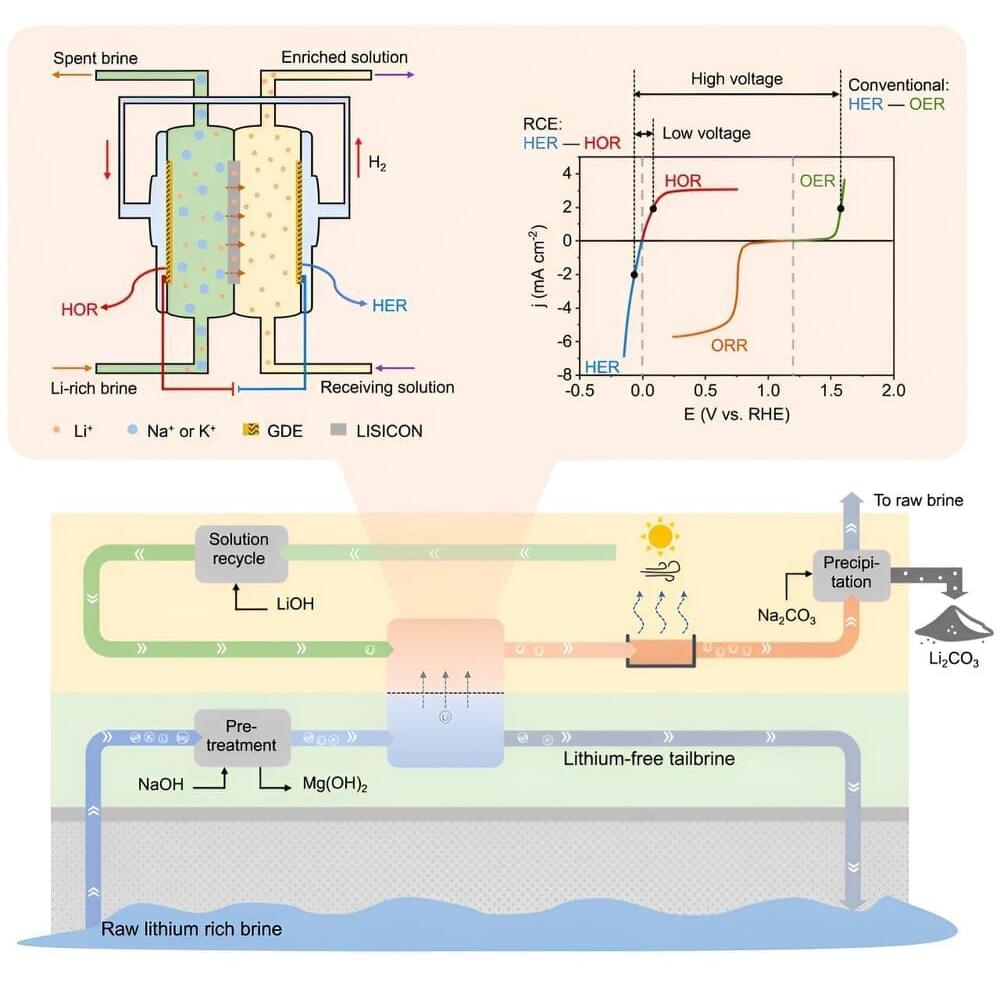
A new technology can extract lithium from brines at an estimated cost of under 40% that of today’s dominant extraction method, and at just a fourth of lithium’s current market price. The new technology would also be much more reliable and sustainable in its use of water, chemicals, and land than today’s technology, according to a study published in Matter by Stanford University researchers.
Global demand for lithium has surged in recent years, driven by the rise of electric vehicles and renewable energy storage. The dominant source of lithium extraction today relies on evaporating brines in huge ponds under the sun for a year or more, leaving behind a lithium-rich solution, after which heavy use of potentially toxic chemicals finishes the job. Water with a high concentration of salts, including lithium, occurs naturally in some lakes, hot springs, and aquifers, and as a byproduct of oil and natural gas operations and of seawater desalination.
Many scientists are searching for less expensive and more efficient, reliable, and environmentally friendly lithium extraction methods. These are generally direct lithium extraction that bypasses big evaporation ponds. The new study reports on the results of a new method using an approach known as “redox-couple electrodialysis,” or RCE, along with cost estimates.