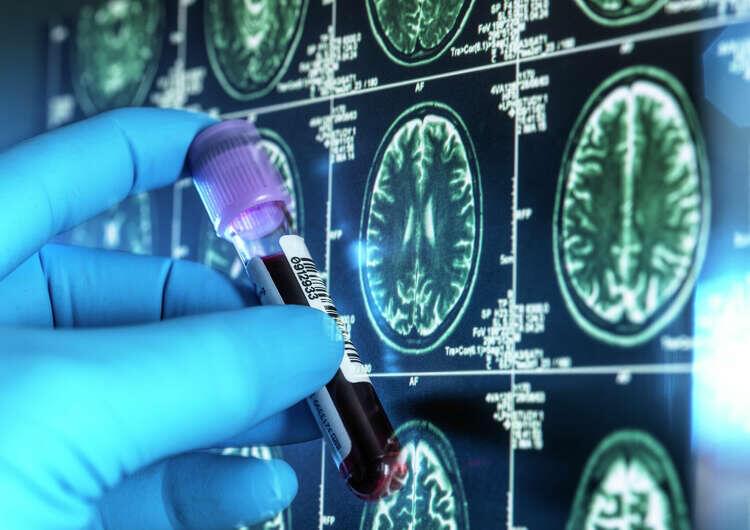
New FDA-approved blood tests for Alzheimer’s offer a faster and less invasive diagnosis, but they’re not approved for everyone. Here’s what to know.
Rice and Baylor researchers have used Rice’s “drug factory” implants to eradicate advanced-stage mesothelioma tumors in mice.

Heat causes errors in the qubits that are the building blocks of a quantum computer, so quantum systems are typically kept inside refrigerators that keep the temperature just above absolute zero (−459 degrees Fahrenheit).
But quantum computers need to communicate with electronics outside the refrigerator, in a room-temperature environment. The metal cables that connect these electronics bring heat into the refrigerator, which has to work even harder and draw extra power to keep the system cold. Plus, more qubits require more cables, so the size of a quantum system is limited by how much heat the fridge can remove.
To overcome this challenge, an interdisciplinary team of MIT researchers has developed a wireless communication system that enables a quantum computer to send and receive data to and from electronics outside the refrigerator using high-speed terahertz waves.
(From 2023)
A new wireless terahertz communication system enables a super-cold quantum computer to send and receive data without generating too much error-causing heat.

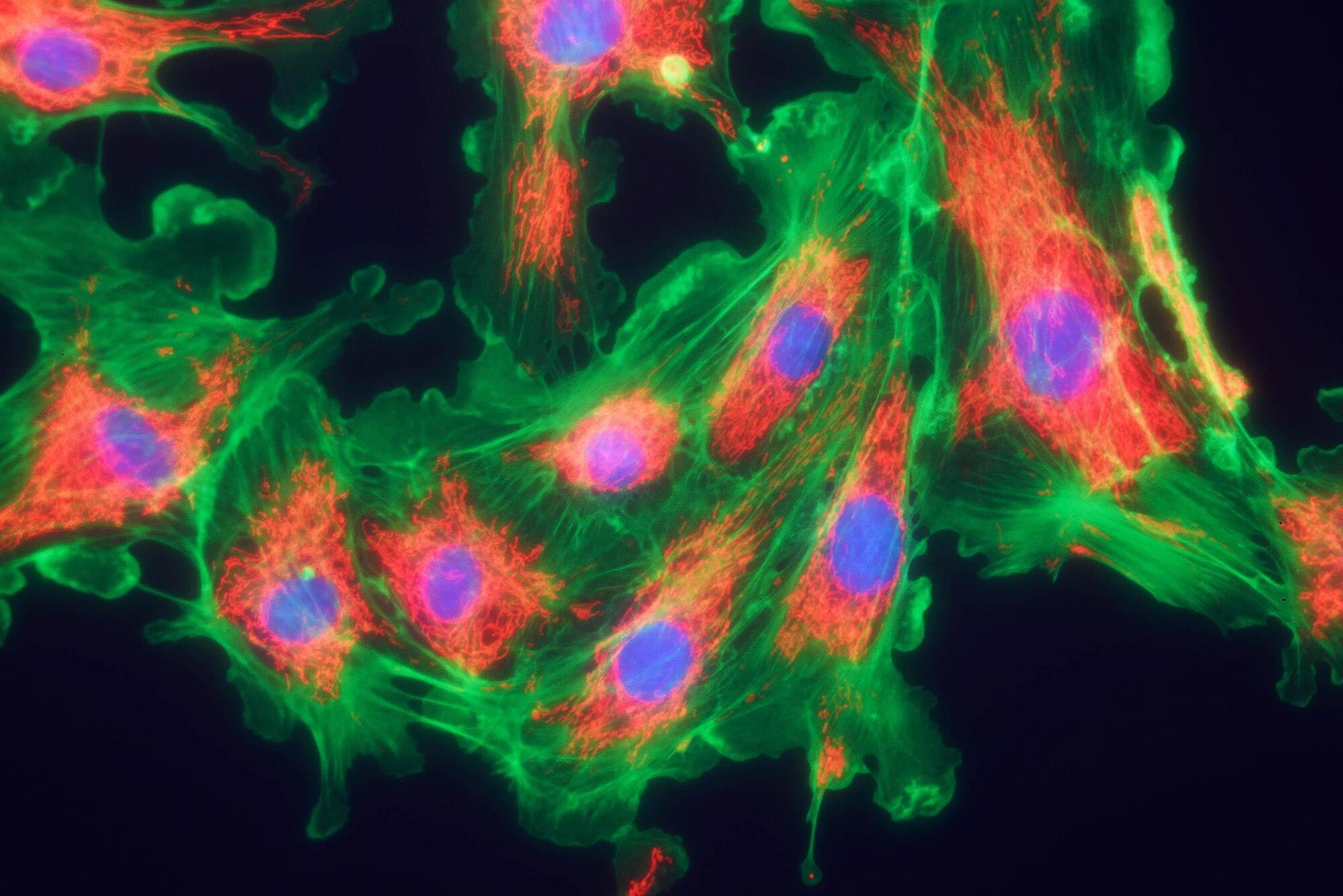
The researchers identified a stress response that emerges from damaged mitochondria. By interrupting this stress response with a compound known as ISRIB, their results showed a marked improvement in blood sugar handling in mice.
A β-cell in the pancreas is responsible for releasing insulin, the hormone that regulates blood sugar.
These cells need high energy output from mitochondria to carry out their job. Weak energy conversion can disrupt insulin release, fueling the symptoms associated with type 2 diabetes.

On 23 June 2025, the world will get a look at the first images from one of the most powerful telescopes ever built: the Vera C. Rubin Observatory.
Perched high in the Chilean Andes, the observatory will take hundreds of images of the southern hemisphere sky, every night for 10 years. In doing so, it will create the most complete time-lapse record of our universe ever assembled. This scientific effort is known as the Legacy Survey of Space and Time (LSST).
Rather than focusing on small patches of sky, the Rubin Observatory will scan the entire visible southern sky every few nights. Scientists will use this rolling deep-sky snapshot to track supernovae (exploding stars), asteroids, black holes, and galaxies as they evolve and change in real time. This is astronomy not as a static snapshot, but as a cosmic story unfolding night by night.

The universe is full of spectacular and violent events, but few are more dramatic than a black hole tearing apart a star. Now, thanks to advanced computer simulations, scientists have gotten their closest look yet at what this cosmic catastrophe might actually look — and even sound — like.
A team of astronomers, led by theoretical astrophysicist Elias Most of the California Institute of Technology (Caltech), modeled the dramatic final milliseconds before a neutron star, the incredibly dense core left behind by a massive stellar explosion, is devoured by a black hole.

IN A NUTSHELL 🤖 Veho has partnered with RIVR to introduce wheeled-legged robots for parcel delivery in Austin, Texas. 🚀 The robots feature precision engineering and adaptive mobility to navigate complex urban environments. 🔗 This collaboration aims to enhance delivery efficiency while reducing physical strain on human drivers. 🌐 The initiative represents a major step

The entire field of mathematics summarised in a single map! This shows how pure mathematics and applied mathematics relate to each other and all of the sub-topics they are made from.
#mathematics #DomainOfScience.
If you would like to buy a poster of this map, they are available here:
North America: https://store.dftba.com/products/map–… else: http://www.redbubble.com/people/domin… French version: https://www.redbubble.com/people/domi… Spanish Version: https://www.redbubble.com/people/domi… I have also made a version available for educational use which you can find here: https://www.flickr.com/photos/9586967… To err is to human, and I human a lot. I always try my best to be as correct as possible, but unfortunately I make mistakes. This is the errata where I correct my silly mistakes. My goal is to one day do a video with no errors! 1. The number one is not a prime number. The definition of a prime number is a number can be divided evenly only by 1, or itself. And it must be a whole number GREATER than 1. (This last bit is the bit I forgot). 2. In the trigonometry section I drew cos(theta) = opposite / adjacent. This is the kind of thing you learn in high school and guess what. I got it wrong! Dummy. It should be cos(theta) = adjacent / hypotenuse. 3. My drawing of dice is slightly wrong. Most dice have their opposite sides adding up to 7, so when I drew 3 and 4 next to each other that is incorrect. 4. I said that the Gödel Incompleteness Theorems implied that mathematics is made up by humans, but that is wrong, just ignore that statement. I have learned more about it now, here is a good video explaining it: • Gödel’s Incompleteness Theorem — Numberphile 5. In the animation about imaginary numbers I drew the real axis as vertical and the imaginary axis as horizontal which is opposite to the conventional way it is done. Thanks so much to my supporters on Patreon. I hope to make money from my videos one day, but I’m not there yet! If you enjoy my videos and would like to help me make more this is the best way and I appreciate it very much.
/ domainofscience Here are links to some of the sources I used in this video. Links: Summary of mathematics: https://en.wikipedia.org/wiki/Mathema… Earliest human counting: http://mathtimeline.weebly.com/early–… First use of zero: https://en.wikipedia.org/wiki/0#History http://www.livescience.com/27853-who–… First use of negative numbers: https://www.quora.com/Who-is-the-inve… Renaissance science: https://en.wikipedia.org/wiki/History… History of complex numbers: http://rossroessler.tripod.com/ https://en.wikipedia.org/wiki/Mathema… Proof that pi is irrational: https://www.quora.com/How-do-you-prov… and https://en.wikipedia.org/wiki/Proof_t… Also, if you enjoyed this video, you will probably like my science books, available in all good books shops around the work and is printed in 16 languages. Links are below or just search for Professor Astro Cat. They are fun children’s books aimed at the age range 7–12. But they are also a hit with adults who want good explanations of science. The books have won awards and the app won a Webby. Frontiers of Space: http://nobrow.net/shop/professor-astr… Atomic Adventure: http://nobrow.net/shop/professor-astr… Intergalactic Activity Book: http://nobrow.net/shop/professor-astr… Solar System App: Find me on twitter, instagram, and my website: http://dominicwalliman.com
/ dominicwalliman
/ dominicwalliman
/ dominicwalliman.
Everywhere else: http://www.redbubble.com/people/domin…
French version: https://www.redbubble.com/people/domi…
Spanish Version: https://www.redbubble.com/people/domi…
I have also made a version available for educational use which you can find here: https://www.flickr.com/photos/9586967…
To err is to human, and I human a lot. I always try my best to be as correct as possible, but unfortunately I make mistakes. This is the errata where I correct my silly mistakes. My goal is to one day do a video with no errors!
1. The number one is not a prime number. The definition of a prime number is a number can be divided evenly only by 1, or itself. And it must be a whole number GREATER than 1. (This last bit is the bit I forgot).