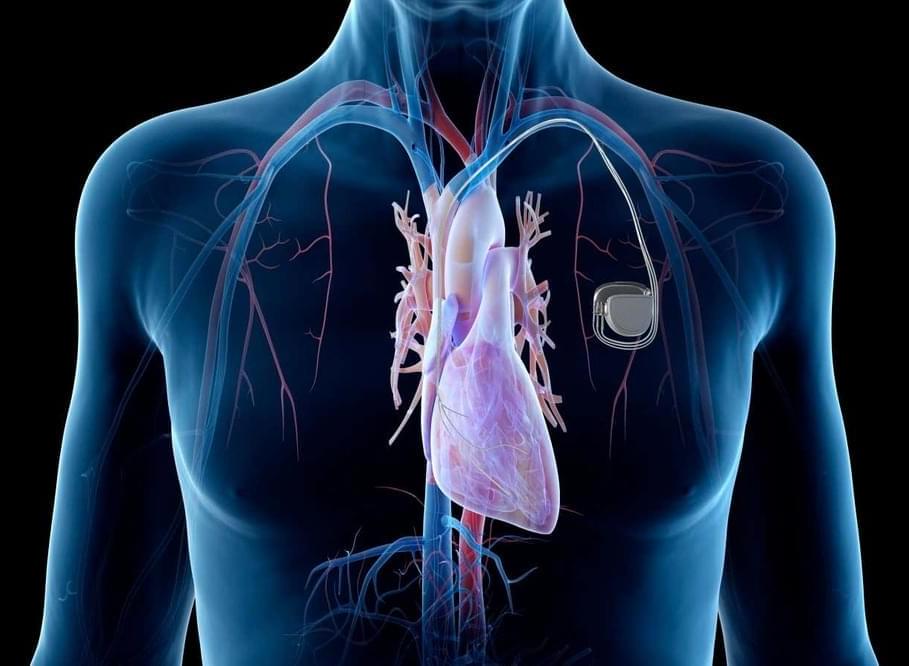
Many medical implants run on batteries that need to be recharged, but what if you could do so just by breathing?

“The increases in THC levels found in cannabis could mimic some of the more pronounced effects that we see for people who are slower metabolizers,” said Dr. Christal Davis.
How can genetics influence cannabis consumption? This is what a recent study published in Addictive Behaviors hopes to address as a team of researchers investigated a link between how genetic variances influence how a person metabolizes THC, which could not only determine future use but also the chances of succumbing to cannabis use disorder, or CUD. This study holds the potential to help cannabis users, medical professionals, legislators, and the public better understand the physiological influences of cannabis use, even at the molecular level.
For the study, the researchers enlisted 54 participants between 18–25 years of age, 38 of whom suffered from CUD while the remaining 16 suffered from non-CUD substance abuse. It has been determined that individuals aged 18–25 have a three times greater likelihood of having CUD compared to individuals over the age of 26. After obtaining blood samples from each participant, the researchers tested them for differences in gene markers, specifically pertaining to THC-metabolizing enzymes. Additionally, each participant was instructed to fill out a questionnaire regarding their experiences with cannabis use and how it makes them feel when they use it.
In the end, the researchers found notable differences between men and women participants, specifically regarding how young women with CUD were found to metabolize THC at slower rates than young women who did not suffer from CUD. For the men, the researchers discovered negative reports from cannabis use with those who also metabolized THC at slower rates, which was the same for both sexes. Additionally, the researchers’ found CUD was more prevalent in individuals who started using cannabis when teenagers, as well. The researchers concluded that proper treatment options for CUD could be proposed due to lack of genetic testing.
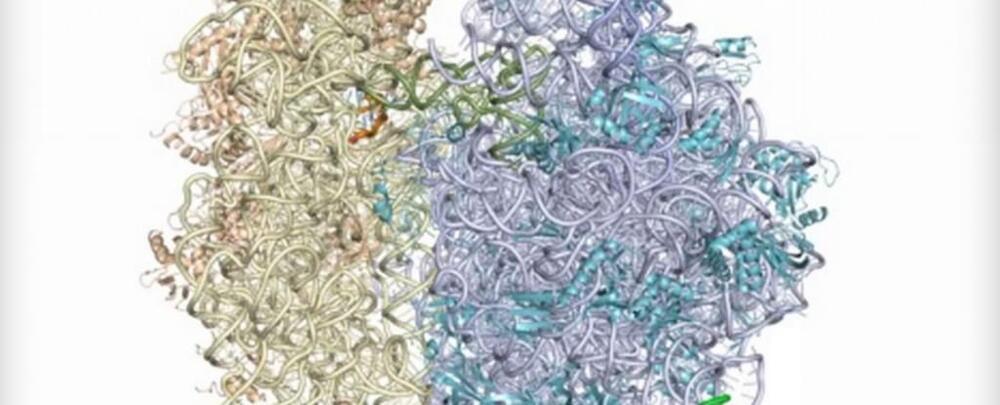
Researchers in the US have developed a synthetic molecular structure called the Ribo-T, and it can be placed inside a living cell to produce specialised proteins and enzymes at almost the same efficiency as an actual ribosome.
Found inside all living cells, ribosomes are dense, complex structures that catalyse a constant stream of protein chains by linking amino acids together in the order specified by messenger RNA (mRNA) molecules. These cellular workhorses are basically in charge of decoding your DNA, and now scientists have manufactured a molecular device that can not only produce protein chains in a test-tube almost as well as a real ribosome, but can also churn out enough protein in bacterial cells without any natural ribosomes to keep them alive.
The team, with researchers from the University of Illinois at Chicago and Northwestern University, says not only will the Ribo-T help them to better understand how our own ribosomes function, but it could lead to more effective drugs and next-gen biomaterials, with these little protein factors churning out whatever we need.
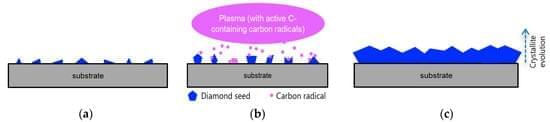
Diamond is a promising material for the biomedical field, mainly due to its set of characteristics such as biocompatibility, strength, and electrical conductivity. Diamond can be synthesised in the laboratory by different methods, is available in the form of plates or films deposited on foreign substrates, and its morphology varies from microcrystalline diamond to ultrananocrystalline diamond. In this review, we summarise some of the most relevant studies regarding the adhesion of cells onto diamond surfaces, the consequent cell growth, and, in some very interesting cases, the differentiation of cells into neurons and oligodendrocytes. We discuss how different morphologies can affect cell adhesion and how surface termination can influence the surface hydrophilicity and consequent attachment of adherent proteins.
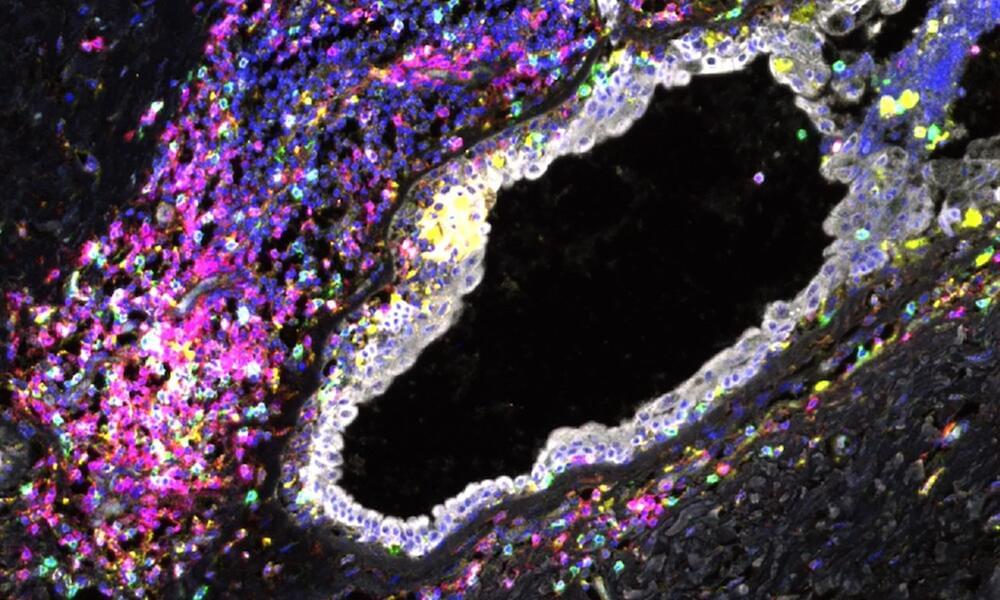
Everyone has BRCA1 and BRCA2 genes, but mutations in these genes—which can be inherited—increase the risk of breast and ovarian cancer.
The study found that the immune cells in breast tissue of healthy women carrying BRCA1 or BRCA2 gene mutations show signs of malfunction known as exhaustion. This suggests that the immune cells can’t clear out damaged breast cells, which can eventually develop into breast cancer.
This is the first time that exhausted immune cells have been reported in non-cancerous breast tissues at such scale—normally these cells are only found in late-stage tumors. The results raise the possibility of using existing immunotherapy drugs as early intervention to prevent breast cancer developing, in carriers of BRCA1 and BRCA2 gene mutations.
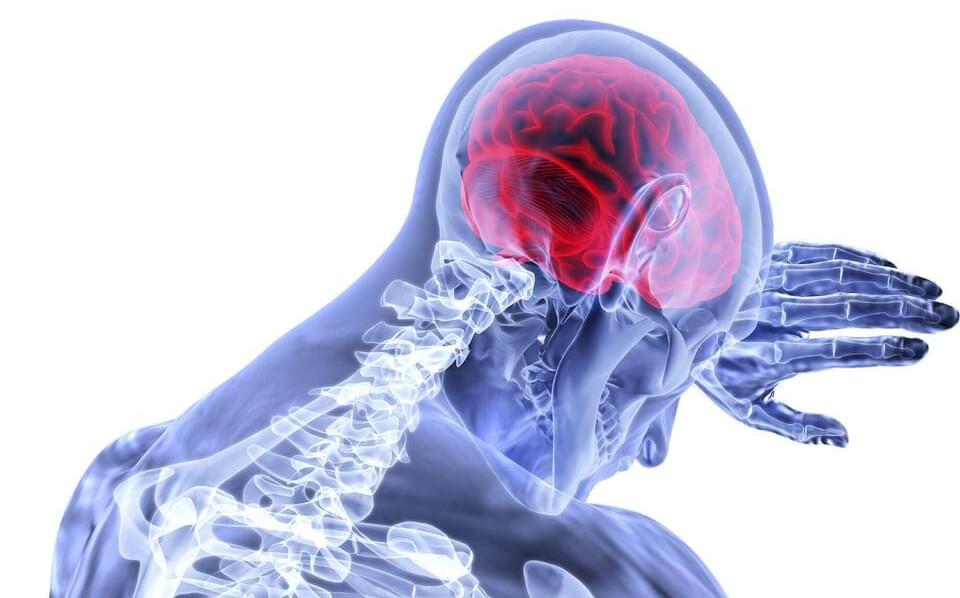
In a new study, AI processed text from health histories and neurologic examinations to locate lesions in the brain. The study, which looked specifically at the large language model called generative pre-trained transformer 4 (GPT-4), is published in the online issue of Neurology Clinical Practice.
A stroke can cause long-term disability or even death. Knowing where a stroke has occurred in the brain helps predict long-term effects such as problems with speech and language or a person’s ability to move part of their body. It can also help determine the best treatment and a person’s overall prognosis.
Damage to the brain tissue from a stroke is called a lesion. A neurologic exam can help locate lesions, when paired with a review of a person’s health history. The exam involves symptom evaluation and thinking and memory tests. People with stroke often have brain scans to locate lesions.

On 28 March it’s the 60th anniversary of the discovery of Epstein-Barr virus, the most common viral infection in humans. The virus was first discovered in association with a rare type of cancer located in Africa, but is now understood to be implicated in 1% of cancers, as well as the autoimmune disease multiple sclerosis. Ian Sample meets Lawrence Young, professor of molecular oncology at Warwick Medical School, to hear the story of this virus, and how it might help us prevent and treat cancer and other illnesses.
{kind=link}