{kind=link}
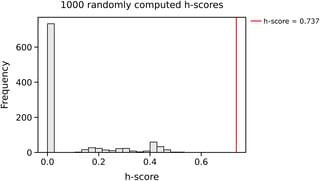
Senescence is a key manifestation of aging at the cellular level, caused by damage incurred by cells in time. In spite of their wide-ranging implications on how our multicellular bodies age, senescent cells are very challenging to identify due to their complex nature: many different aspects of cells are affected by this cellular state. This complicates defining clear criteria that help us decide whether a cell is senescent or not. In this paper, we propose a computational pipeline that enables us to identify a small subset of genes associated with senescence. The method combines two approaches commonly used in the study of networks, community detection and node centrality, and applies them to gene expression data obtained from the muscle tissue of mice after damage. The results obtained can contribute to establish the molecular correlates of a complex cellular state such as senescence.
Citation: Sabalic A, Moiseeva V, Cisneros A, Deryagin O, Perdiguero E, Muñoz-Cánoves P, et al. (2026) Cell-type resolved transcriptional network analysis of in vivo cellular senescence following injury. PLoS Comput Biol 22: e1014429. https://doi.org/10.1371/journal.pcbi.
Editor: Christoph Kaleta, Christian Albrechts Universitat zu Kiel, GERMANY.