Aging human breast atlas reveals cancer susceptibility
The team used advanced imagining techniques to analyse breast tissue from more than 500 women aged 15 to 86 years old. The tissue included biopsies taken from women for non-cancer-related reasons.
Combining these images with details of the hormone receptors and immune cells present, as well as the tissue architecture, the researchers were able to map how breast tissue changes over time in unprecedented detail. Their findings point to reasons why breast cancer risk increases with age and why tumors in younger women differ biologically.
The author added: “Our map revealed that as women age, their breast tissue goes through major changes, with the most dramatic changes occurring at menopause. There are changes, too, during their twenties, possibly linked to pregnancy and childbirth, but these are far less pronounced.”
The map revealed that all types of cells become fewer in number and divide far less often. Milk-producing structures known as lobules shrink or disappear, while the ducts that that carry milk become relatively more common, with the supporting layer around them becoming thicker. Fat cells increase while blood vessels decrease.
Meanwhile, changes occur in the immune environment. Younger breasts have more B cells and active T cells, which helps them identify and kill cancer cells. As tissue ages, these types of cells decline in number, replaced by other types of immune cell that indicate a more inflammatory and potentially less protective immune environment. ScienceMission sciencenewshighlights.
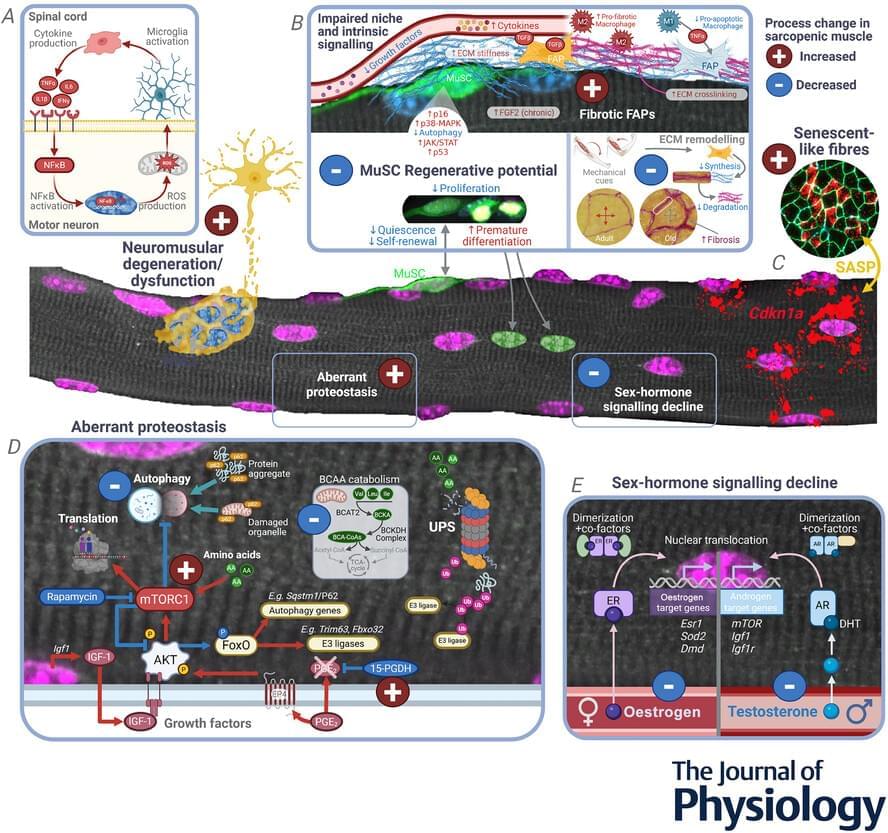
The exchange of cellular and molecular cues between motor neurons and skeletal muscle is required for sustaining muscle mass and function throughout life (Shi et al., 2012). Loss of neural input to skeletal muscle, whether gradual due to sarcopenia or catastrophic due to neurodegenerative disease (e.g. amyotrophic lateral sclerosis, ALS), results in neurogenic muscle atrophy (Brown & Al-Chalabi, 2017 ; Sarto et al., 2024). In humans, the number of motor neurons in the lumbosacral cord remains stable until 60 years of age, after which numbers can decline up to 50% compared to middle age. In healthy ageing, motor neuron loss is compensated for by robust reinnervation of denervated fibres (Piasecki et al., 2018). In sarcopenia, the capacity for re-innervation is lost over time, resulting in permanent denervation of muscle fibres and a reduction in the number and diameter of ventral root motor axons (Kawamura et al., 1977 ; Mittal & Logmani, 1987). In rodents and humans, this loss of capacity for reinnervation also leads to a loss of motor unit potential and reduced muscle mass and function (Aare et al., 2016 ; Chai et al., 2011 ; Horwath et al., 2025 ; Kung et al., 2014 ; Piasecki et al., 2018 ; Rowan et al., 2012).
Though the drivers of neuronal degeneration are best exemplified in conditions like ALS, they are equally relevant for sarcopenia (Azzolino et al., 2024). A key pathogenic mechanism of neuronal degeneration is hyperexcitability of motor neurons in the motor cortex (Menon et al., 2020 ; Vucic et al., 2008). However, evidence from ALS rodent models suggests that motor neurons in the spinal cord exhibit a spectrum of phenotypes ranging from hypoexcitability (Filipchuk et al., 2021 ; Martinez-Silva et al., 2018) to hyperexcitability (Pambo-Pambo et al., 2009). While hypoexcitability results in loss of repetitive firing and a breakdown in synaptic communication (Martinez-Silva et al., 2018), hyperexcitability promotes excessive glutamatergic signalling and intracellular calcium overload, thereby triggering neuronal degeneration (Shaw & Ince, 1997). Although hyperexcitability is not a common feature of sarcopenia, reduced excitability of motor neurons in the motor cortex and spinal cord has been linked to lower muscle strength, smaller motor evoked potentials and reduced functional capacity in sarcopenic older adults (Clark et al., 2015, 2021 ; Orssatto et al., 2025).
Other factors contributing to motor neuron death include oxidative stress, mitochondrial dysfunction and inflammation, which form interlinked, self-reinforcing cycles in which oxidative stress drives mitochondrial dysfunction, amplifying oxidative stress, inflammation and neuronal death (Xu et al., 2025). Increased oxidative damage, protein nitration and lipid oxidation are observed in the motor neurons and neuronal tissue of the copper/zinc superoxide dismutase-deficient (Sod1−/−) mouse, a model of increased oxidative stress that displays accelerated sarcopenia phenotypes (Deepa et al., 2019 ; Sims-Robinson et al., 2013). Synapses exposed to excessive reactive oxygen species (ROS) exhibit reduced binding of glutamate to its transporters (Rao et al., 2003 ; Volterra et al., 1994). The resultant increase in glutamate at the synapse promotes excitotoxicity and calcium accumulation in motor neurons. This calcium can enter mitochondria, impairing mitochondrial function and further increasing the production of ROS (Pivovarova & Andrews, 2010). Mitochondrial dysfunction is also an archetypal feature of sarcopenia. A study of post-mortem spinal cord tissue from 68-to 99-year-olds showed evidence of marked mitochondrial dysfunction (Rygiel et al., 2014), with extensive consequences ranging from bioenergetic failure through to chronic inflammation. With respect to the latter, excessive ROS that arises from dysfunctional mitochondria can modulate innate immunity through redox-sensitive inflammatory pathways, including nuclear factor-κB (NF-κB; Morris et al., 2022), or directly trigger the inflammasome, leading to activation of caspase-1 and subsequent cleavage and activation of potent pro-inflammatory cytokines (e.g. interleukin (IL)-1β and IL-18) (Cerretti et al., 1992 ; Keller et al., 2008). Not surprisingly, inflammation and progressively dysfunctional microglia and astrocytes are commonly reported in ageing (Beers & Appel, 2019 ; Castro et al., 2024 ; Piekarz et al., 2020 ; Troost et al., 1990 ; von Bernhardi & Eugenín, 2025).