This study describes a novel ATXN2 expansion within the classic pathogenic range for spinocerebellar ataxia 2 that manifests as an early-onset neurodegenerative disorder in the homozygous state, while being asymptomatic into late adulthood in the heterozygous state.
The length and content of ATXN2 trinucleotide repeat significantly influences disease development and clinical phenotype. ATXN2 alleles containing 13–31 CAG trinucleotide repeats are normal and commonly found in healthy individuals4 and over 90% of tested individuals possess an allele containing 22 CAG repeats.21 Spinocerebellar ataxia type 2 is caused by dominant alleles of 33 or more CAG trinucleotide repeats.11,22 Alleles containing 33–34 CAG repeats are considered reduced penetrance alleles, and carriers may or may not develop late onset ataxia.22 Fully penetrant alleles most commonly have 37–39 CAG repeats and are pathogenic for SCA2.11 While SCA2 alleles of 31 pure CAG repeats exhibit high instability on inheritance, it has been proposed that CAA interruptions confers meiotic stability.23 An anticipation phenomenon in SCA2 has also been described, consisting of earlier disease onset and increased clinical severity in subsequent generations which are mirrored by an increase in CAG repeat size.12 Patients with SCA2-related parkinsonism carry intermediate range alleles and possess alleles with CAA interruptions.24,25 Similarly, ATNX2 variants associated with ALS are CAA interrupted and are rarely in the pathogenic range of SCA2.26,27 Contrasting with trinucleotide expansion diseases, repeat size has no bearing on ALS AO but correlates with disease risk.28 ATXN2 has been identified as a disease modifier gene for a variety of neurologic conditions and similarly, various genes may influence the AO of SCA2, including long normal repeats in the CACNA1A and RAI1 genes.29 Nonetheless, the most important predictor of AO and clinical severity remains the polyglutamine repeat expansion size.30
Infantile and childhood forms of SCA2 are described, and these patients present with a multi-systematic neurodegenerative disorder including developmental delay, retinitis pigmentosa, optic atrophy, hypotonia, seizures, facial dysmorphism, dystonic features, and early mortality.21,31 Infantile cases all possess extreme length CAG repeats (range 69–884) in the heterozygous state, with clinical severity related to repeat size, and inherited with an anticipation phenomenon from parents within the fully penetrant range of SCA2 (range 39–47 CAG repeats).21,31
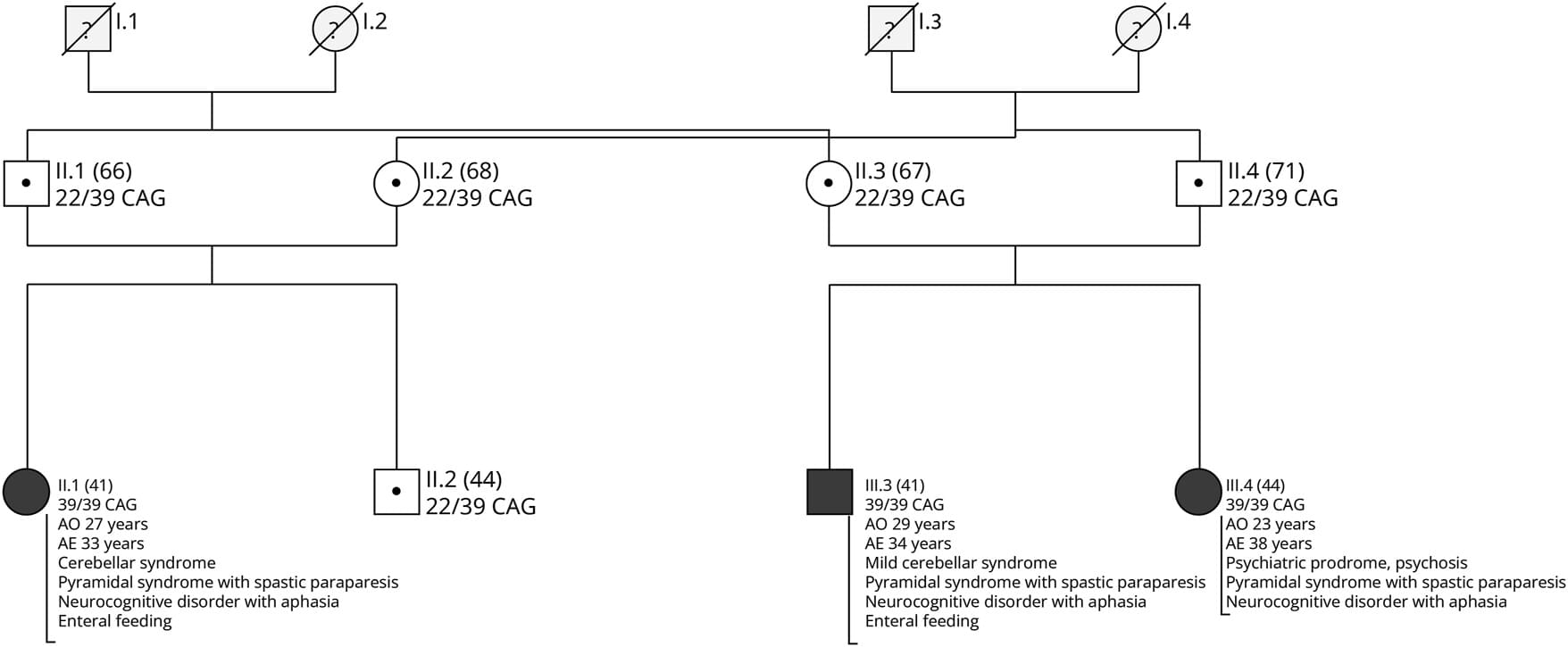
Homozygous cases of SCA2 are exceedingly rare.32,33 Notably, a patient with 31/31 CAG alleles developed late-onset cerebellar ataxia, suggesting that patients with homozygous variants may manifest signs of disease within a nonpathogenic variant range, that is not associated with disease development in the heterozygous state.18,32 Two homozygous cases from an Indian family with 35/37 and 36/39 CAG repeats alleles developed early onset, levodopa responsive Parkinson disease without ataxia,33 while several family members with heterozygous ATXN2 variants exhibited parkinsonism and/or ataxia with variable ages of onset ranging from adulthood to their sixties.33 Moreover, two homozygous cases with intermediate alleles of 32/3217 and 33/3327 displayed a pure ALS phenotype, without ataxia. These cases highlight the phenotypical variability of homozygous ATXN2 variants.