This study broadens the phenotypic and genetic spectrum of PNH, demonstrating a dual PNH phenotype associated with a bi‑transcript mechanism and mosaic inheritance, including tissue‑specific mosaicism.
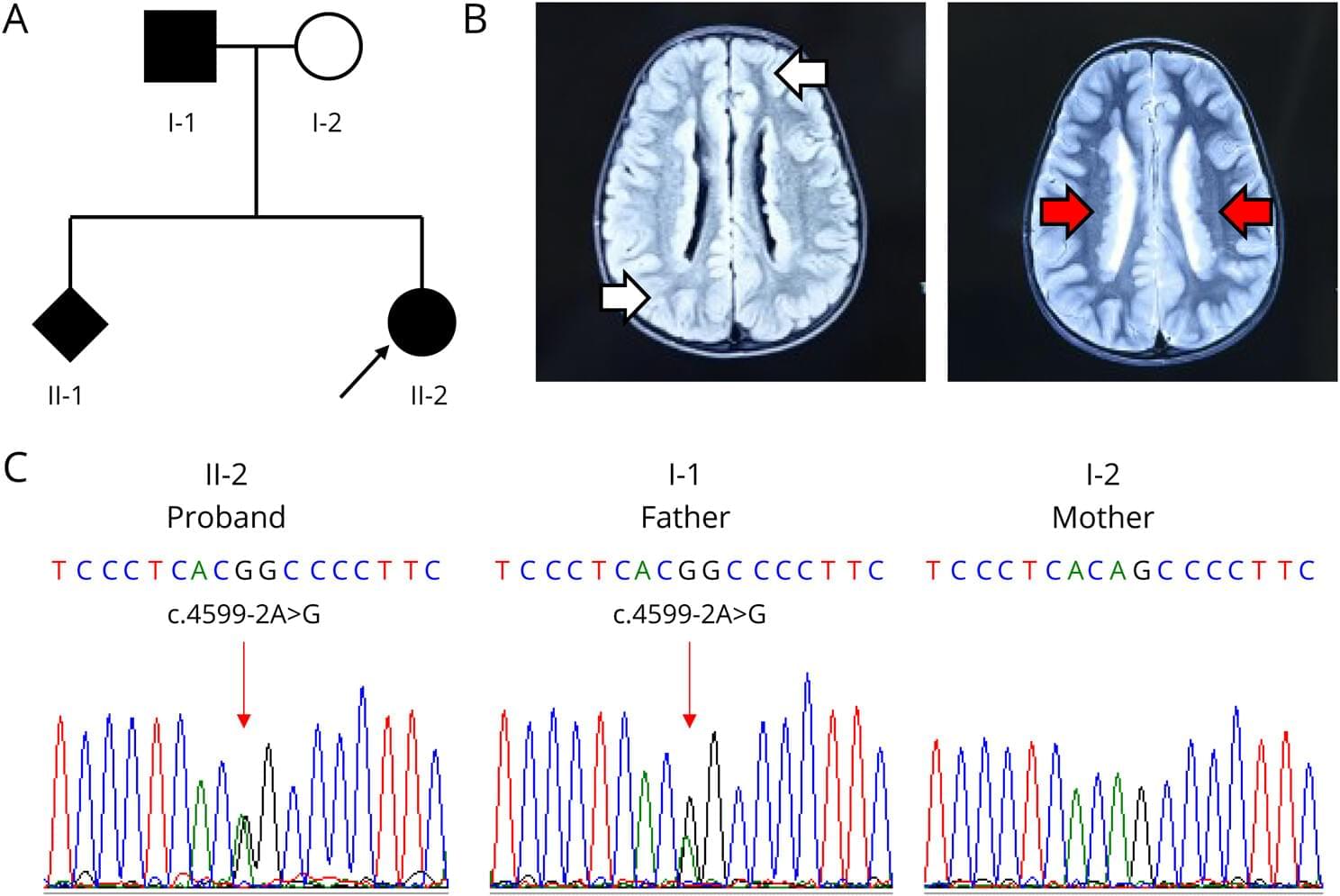
PNH is a neurodevelopmental brain malformation characterized by failure of the gray matter to properly migrate to the cerebral cortex during embryonic development. This results in ectopic localization around the ventricular ependyma.1 MRI serves as the primary diagnostic tool, showing bilateral periventricular gray matter nodules with a signal intensity similar to that of normal cortical gray matter.2,3 Its primary clinical manifestation is epilepsy, which is often accompanied by intellectual disabilities and learning difficulties.2,3 PNH is genetically heterogeneous and is linked to variants in multiple genes, including ARFGEF2, ERMARD, NEDD4L, ARF1, and MAP1B, as well as abnormalities in chromosome 5. Among these, pathogenic variants in FLNA are the most common genetic causes.4
FLNA is located at Xq28 and comprises 47 exons,5 encoding a 280 kDa actin binding protein, called filamin A. The N-terminal region contains an actin binding domain (ABD) and a rod-like structure composed of 24 immunoglobulin-like repeats. ABD interacts with actin to stabilize the cytoskeletal architecture and plays crucial roles in maintaining cell shape, migration, and transmitting mechanical force. FLNA regulates cellular migration and extension processes via interactions with several signaling proteins, including small GTPases Rac/Rho, TRAF2, integrins, and BRCA2.6–8 This gene possesses at least 2 transcription initiation sites (ENST00000369850.8 and ENST00000610817.4) that use distinct promoters and demonstrate tissue-specific expression.6–8 Rat FLNA-knockdown models exhibit impaired neuronal migration and elevated epileptic susceptibility.