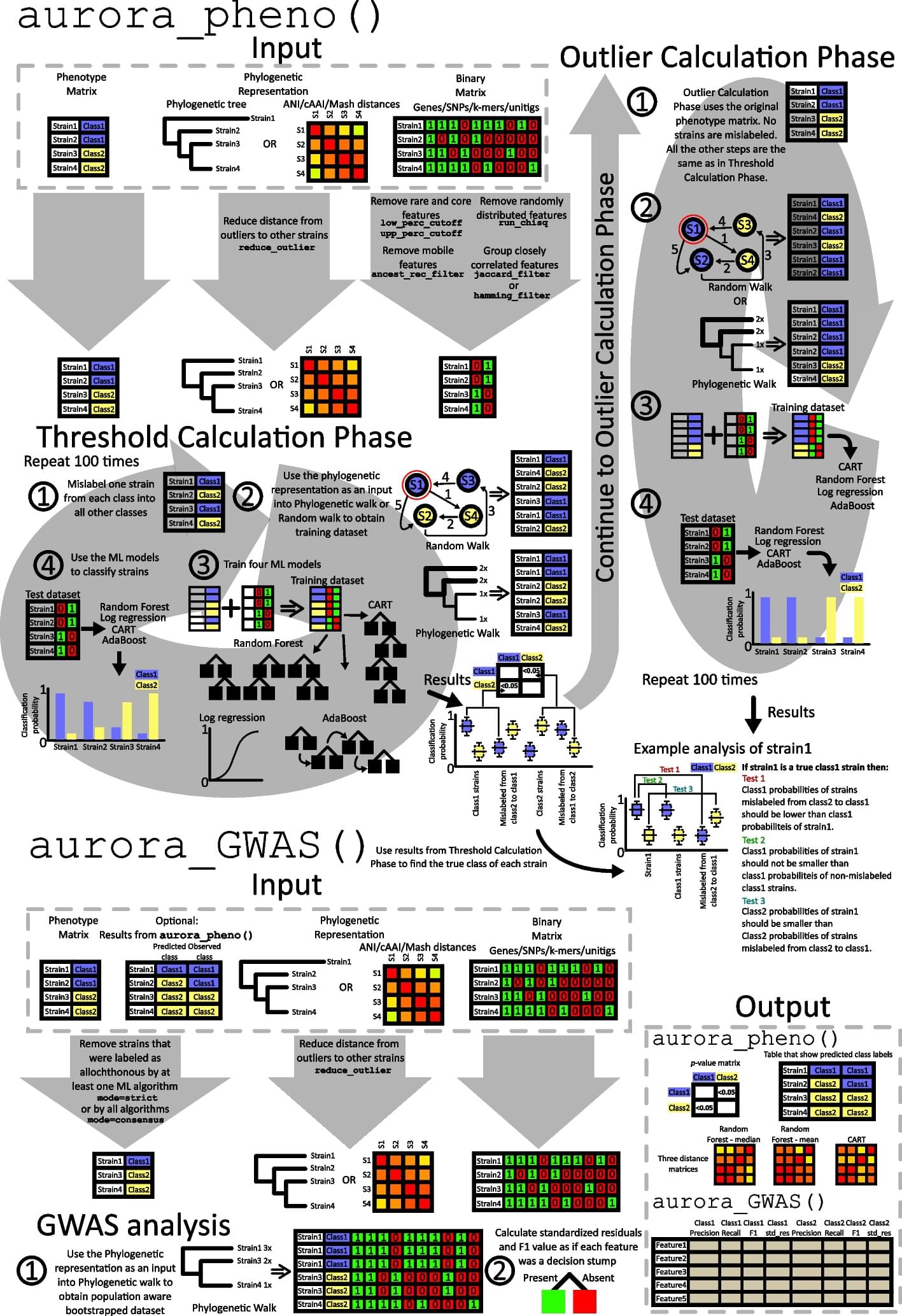
A primary goal of microbial genome-wide association studies is identifying genomic variants associated with a particular habitat. Existing tools fail to identify known causal variants if the analyzed trait shaped the phylogeny. Furthermore, due to inclusion of allochthonous strains or metadata errors, the stated sources of strains in public databases are often incorrect, and strains may not be adapted to the habitat from which they were isolated. We describe a new tool, aurora, that identifies autochthonous strains and the genes associated with habitats while acknowledging the potential role of the habitat adaptation trait in shaping phylogeny.