Researchers at the Fritz Haber Institute have developed the Automatic Process Explorer (APE), an approach that enhances our understanding of atomic and molecular processes. By dynamically refining simulations, APE has uncovered unexpected complexities in the oxidation of palladium (Pd) surfaces, offering new insights into catalyst behavior. The study is published in the journal Physical Review Letters.
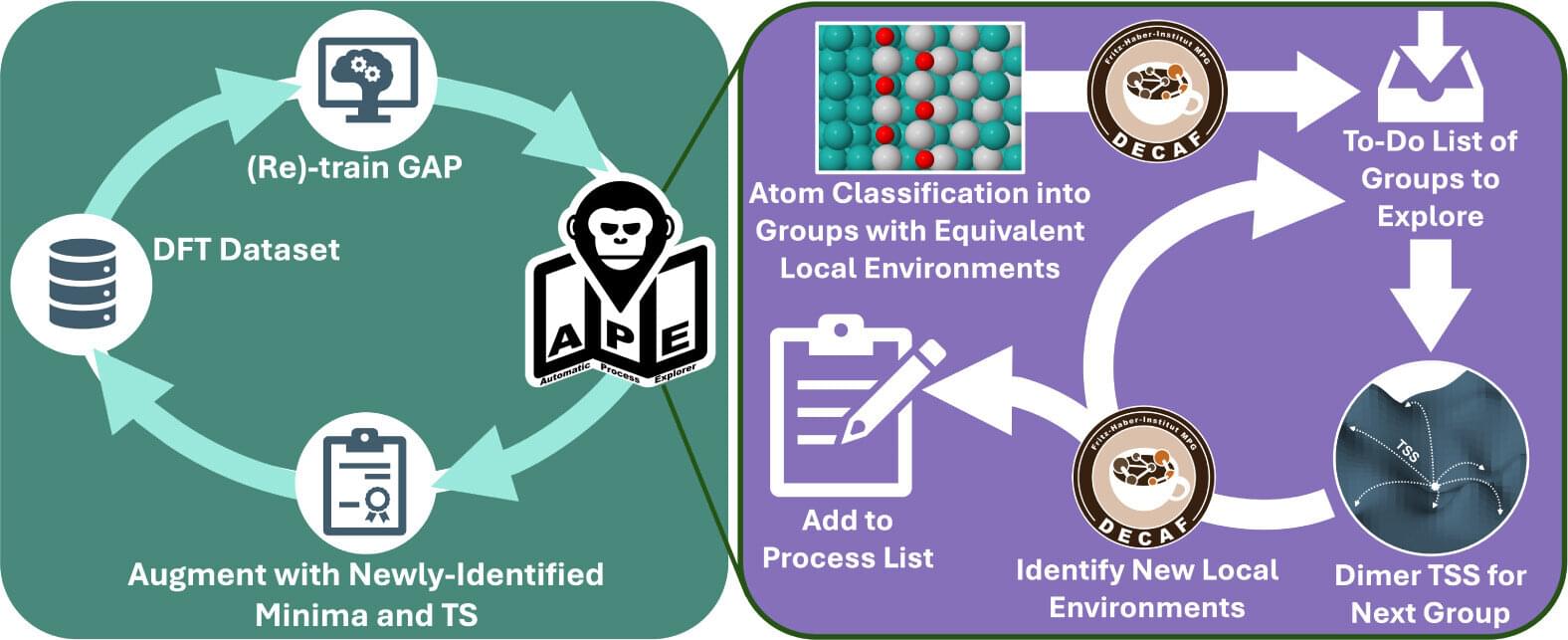
Kinetic Monte Carlo (kMC) simulations are essential for studying the long-term evolution of atomic and molecular processes. They are widely used in fields like surface catalysis, where reactions on material surfaces are crucial for developing efficient catalysts that accelerate reactions in energy production and pollution control. Traditional kMC simulations rely on predefined inputs, which can limit their ability to capture complex atomic movements. This is where the Automatic Process Explorer (APE) comes in.
Developed by the Theory Department at the Fritz Haber Institute, APE overcomes biases in traditional kMC simulations by dynamically updating the list of processes based on the system’s current state. This approach encourages exploration of new structures, promoting diversity and efficiency in structural exploration. APE separates process exploration from kMC simulations, using fuzzy machine-learning classification to identify distinct atomic environments. This allows for a broader exploration of potential atomic movements.