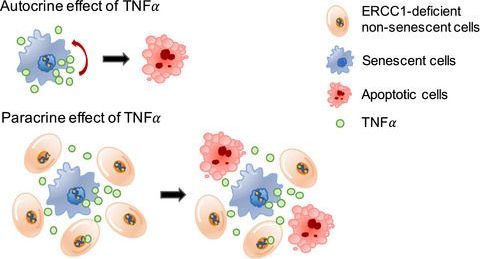
ERCC1 (excision repair cross complementing‐group 1) is a mammalian endonuclease that incises the damaged strand of DNA during nucleotide excision repair and interstrand cross‐link repair. Ercc1−/Δ mice, carrying one null and one hypomorphic Ercc1 allele, have been widely used to study aging due to accelerated aging phenotypes in numerous organs and their shortened lifespan. Ercc1−/Δ mice display combined features of human progeroid and cancer‐prone syndromes. Although several studies report cellular senescence and apoptosis associated with the premature aging of Ercc1−/Δ mice, the link between these two processes and their physiological relevance in the phenotypes of Ercc1−/Δ mice are incompletely understood. Here, we show that ERCC1 depletion, both in cultured human fibroblasts and the skin of Ercc1−/Δ mice, initially induces cellular senescence and, importantly, increased expression of several SASP (senescence‐associated secretory phenotype) factors. Cellular senescence induced by ERCC1 deficiency was dependent on activity of the p53 tumor‐suppressor protein. In turn, TNFα secreted by senescent cells induced apoptosis, not only in neighboring ERCC1‐deficient nonsenescent cells, but also cell autonomously in the senescent cells themselves. In addition, expression of the stem cell markers p63 and Lgr6 was significantly decreased in Ercc1−/Δ mouse skin, where the apoptotic cells are localized, compared to age‐matched wild‐type skin, possibly due to the apoptosis of stem cells. These data suggest that ERCC1‐depleted cells become susceptible to apoptosis via TNFα secreted from neighboring senescent cells. We speculate that parts of the premature aging phenotypes and shortened health‐ or lifespan may be due to stem cell depletion through apoptosis promoted by senescent cells.