{kind=link}
Aging manifests itself through a decline in organismal homeostasis and a multitude of cellular and physiological functions. Efforts to identify a common basis for vertebrate aging face many challenges; for example, while there have been documented changes in the expression of many hundreds of mRNAs, the results across tissues and species have been inconsistent. We therefore analyzed age-resolved transcriptomic data from 17 mouse organs and 51 human organs using unsupervised machine learning3 –5 to identify the architectural and regulatory characteristics most informative on the differential expression of genes with age. We report a hitherto unknown phenomenon, a systemic age-dependent length-driven transcriptome imbalance that for older organisms disrupts the homeostatic balance between short and long transcript molecules for mice, rats, killifishes, and humans. We also demonstrate that in a mouse model of healthy aging, length-driven transcriptome imbalance correlates with changes in expression of splicing factor proline and glutamine rich (Sfpq), which regulates transcriptional elongation according to gene length. Furthermore, we demonstrate that length-driven transcriptome imbalance can be triggered by environmental hazards and pathogens. Our findings reinforce the picture of aging as a systemic homeostasis breakdown and suggest a promising explanation for why diverse insults affect multiple age-dependent phenotypes in a similar manner.
The transcriptome responds rapidly, selectively, strongly, and reproducibly to a wide variety of molecular and physiological insults experienced by an organism. While the transcripts of thousands of genes have been reported to change with age, the magnitude by which most transcripts change is small in comparison with classical examples of gene regulation2,8 and there is little consensus among different studies. We hence hypothesize that aging is associated with a hitherto uncharacterized process that affects the transcriptome in a systemic manner. We predict that such a process could integrate heterogenous, and molecularly distinctive, environmental insults to promote phenotypic manifestations of aging.
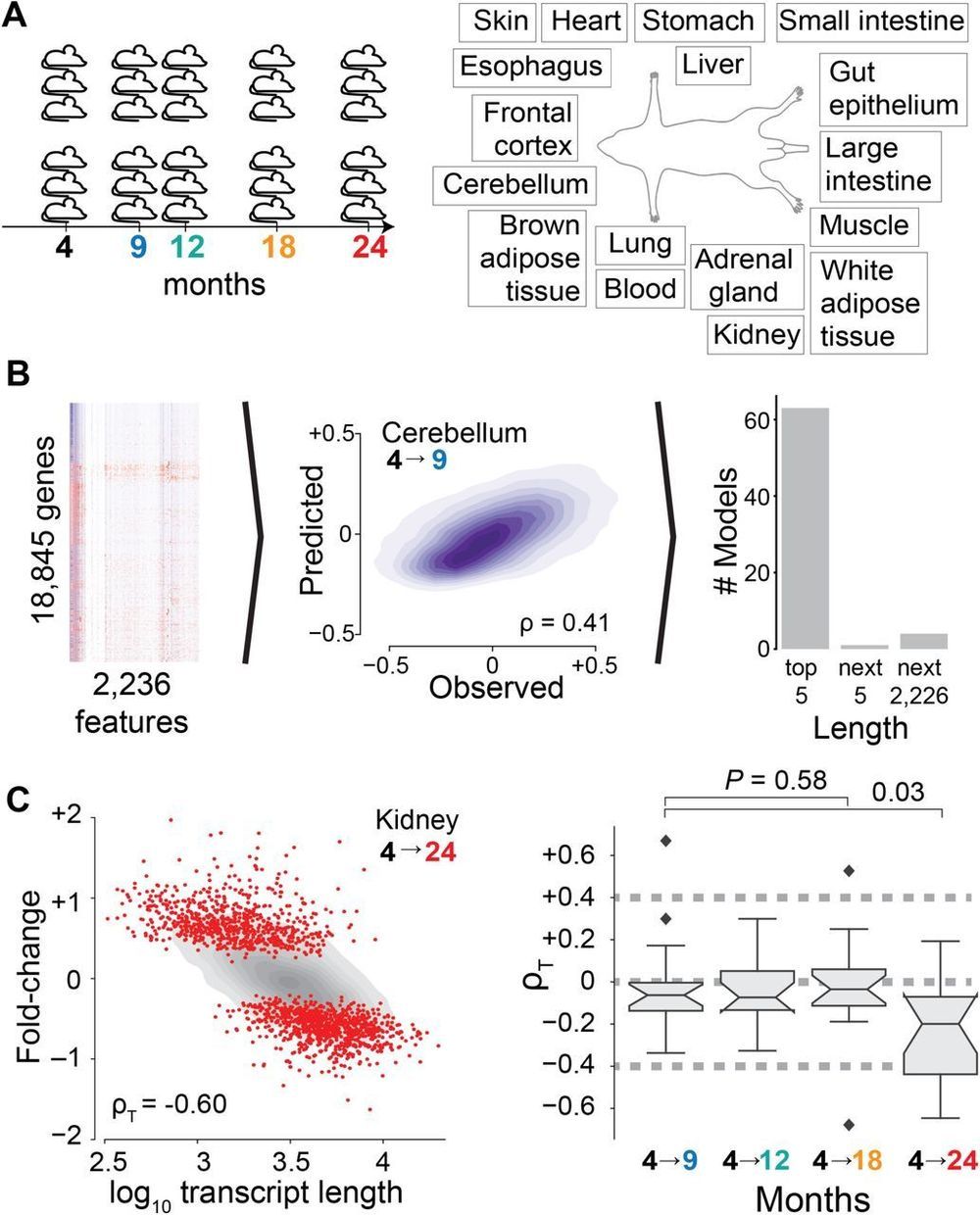
We use an unsupervised machine learning approach3 –5 to identify the sources of age-dependent changes in the transcriptome. To this end, we measure and survey the transcriptome of 17 mouse organs from 6 biological replicates at 5 different ages from 4 to 24 months raised under standardized conditions (Fig. 1A). We consider information on the structural architecture of individual genes and transcripts, and knowledge on the binding of regulatory molecules such as transcription factors and microRNAs (miRNAs) (Fig. 1B). We define age-dependent fold-changes as the log2-transformed ratio of transcripts of one gene at a given age relative to the transcripts of that gene in the organs of 4-month-old mice. As expected for models capturing most measurable changes in transcript abundance, the predicted fold-changes (Fig. S1) match changes empirically observed between distinct replicate cohorts of mice (Figs. S2 and S3).