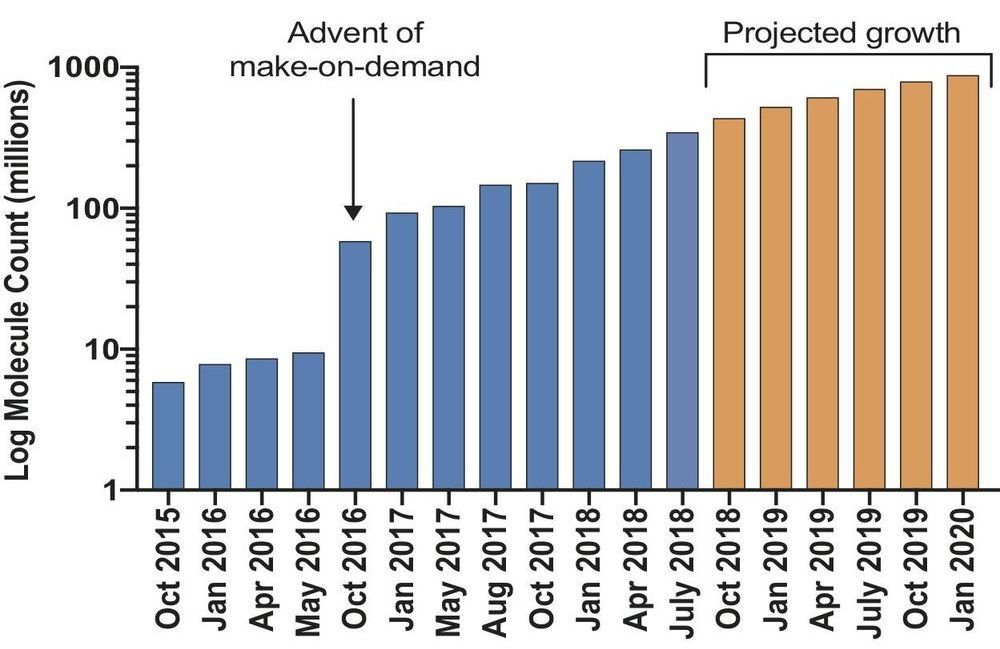
Researchers have launched an ultra-large virtual docking library expected to grow to more than 1 billion molecules by next year. It will expand by 1000-fold the number of such “make-on-demand” compounds readily available to scientists for chemical biology and drug discovery. The larger the library, the better its odds of weeding out inactive “decoy” molecules that could otherwise lead researchers down blind alleys. The project is funded by the National Institutes of Health.
“To improve medications for mental illnesses, we need to screen huge numbers of potentially therapeutic molecules,” explained Joshua A. Gordon, M.D., Ph.D., director of NIH’s National Institute of Mental Health (NIMH), which co-funded the research. “Unbiased computational modeling allows us to do this in a computer, vastly expediting the process of discovering new treatments. It enables researchers to virtually “see” a molecule docking with its receptor protein—like a ship in its harbor berth or a key in its lock—and predict its pharmacological properties, based on how the molecular structures are predicted to interact. Only those relatively few candidate molecules that best match the target profile on the computer need to be physically made and tested in a wet lab.”
Bryan Roth, M.D., Ph.D., of the University of North Carolina (UNC) Chapel Hill, Brian Shoichet, Ph.D., and John Irwin, Ph.D., of the University of California San Francisco, and colleagues, report on their findings Feb. 6, 2019 in the journal Nature. The study was supported, in part, by grants from NIMH, National Institute of General Medical Sciences (NIGMS), the NIH Common Fund, and National Institute of Neurological Disorders and Stroke (NINDS).
Read more