Even as cells split, the genome quietly holds on to its structure and its memory.






Through new experiments, researchers in Japan and Germany have recreated the chemical conditions found in the subsurface ocean of Saturn’s moon, Enceladus. Published in Icarus, the results show that these conditions can readily produce many of the organic compounds observed by the Cassini mission, strengthening evidence that the distant world could harbor the molecular building blocks of life.
Beneath its thick outer shell of ice, astronomers widely predict that Saturn’s sixth largest moon hosts an ocean of liquid water in its south polar region. The main evidence for this ocean is a water-rich plume which frequently erupts from fractures in Enceladus’ surface, leaving a trail of ice particles in its orbital paths which contributes to one of its host planet’s iconic rings.
Between 2004 and 2017, NASA’s Cassini probe passed through this E-ring and plume several times. Equipped with instruments including mass spectrometers and an ultraviolet imaging spectrograph, it detected a diverse array of organic compounds: from simple carbon dioxide to larger hydrocarbon chains, which on Earth are essential molecular precursors to complex biomolecules.
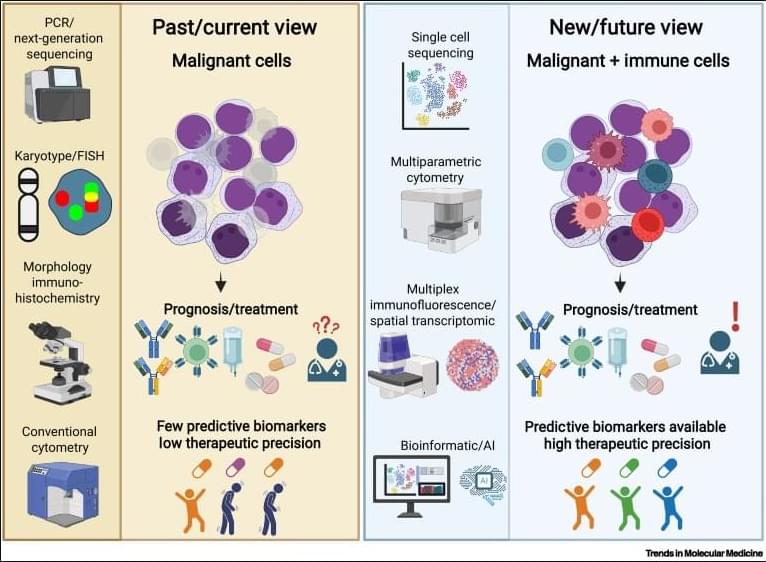
Why immunoscores work in solid tumors—but not yet in blood cancers👇
✅In solid tumors, immune profiling has reached a high level of standardization. Clear tumor boundaries allow quantification of immune cell infiltration, particularly CD3⁺ and CD8⁺ T cells, using immunohistochemistry. This has led to the development of validated immunoscores that stratify tumors as “hot,” “cold,” or “very cold,” providing robust prognostic and predictive value for immunotherapy response.
✅These immunoscores work because solid tumors are spatially organized. Immune cells can be classified as infiltrating or excluded, and their density within defined tumor regions directly correlates with clinical outcome. As a result, immune cell infiltration has become a reliable biomarker to guide treatment decisions in cancers such as colon carcinoma.
✅In contrast, hematologic malignancies lack these defining features. Leukemias and lymphomas are systemic diseases without clear tumor borders, making spatial immune assessment fundamentally challenging. Malignant and nonmalignant immune cells coexist within the same compartments, blurring the distinction between tumor cells and the immune microenvironment.
✅Current immune profiling in hematologic cancers relies on baseline physiological levels of circulating or tissue-resident immune cells, including monocytes, neutrophils, T cells, NK cells, and B cells. While techniques such as flow cytometry, histology, and bulk or single-cell RNA sequencing provide rich datasets, they do not yet translate into a unified, clinically actionable immune score.
✅This lack of standardization creates uncertainty in predicting immunotherapy responses. Metrics such as inflammation, cytotoxicity, or immune infiltration are difficult to interpret consistently across patients and disease subtypes, especially given systemic involvement and tissue-specific immune contexts.
💡
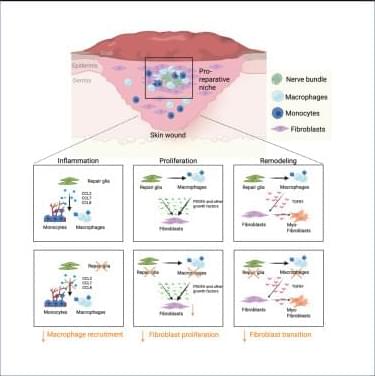
Tissue repair involves extensive communication between the different cellular components of the skin. Among them, nerve innervation is critical for a successful repair process.12,13,14,55 However, only in recent years has the pro-reparative contribution of peripheral glial cells been acknowledged.56,57 For instance, peripheral glia support progenitor cell proliferation by secreting growth factors such as newt anterior gradient protein in the amphibian blastema58 and oncostatin M (OSM) and PDGFα in the digit tip blastema.49 Previous work from our group and others has shown that peripheral glial cells promote skin repair, as depletion of these cells decreased dermal and epidermal cell proliferation,49 reduced myofibroblast numbers,18 and, ultimately, impaired skin wound healing.
Here, we found that peripheral glial cells, primarily residing in NBs, constitute a pro-reparative niche, enriched in inflammatory cells, fibroblasts, and high cell proliferation, essential for the healing process of acute skin wounds. Pro-reparative niches have previously been described in the skin epithelium and in the skeletal muscle, where local stem cell microenvironments support tissue homeostasis.59 In addition, non-myelinating glial cells were shown to be part of a stem cell niche sustaining hemopoietic stem cell dormancy by secreting TGF-β.60 Moreover, enteric glial cells were recently identified to regulate intestinal stem cell turnover by secreting wingless int-1 (WNTs) and were shown to envelop the intestinal stem cells by forming a web-like structure around the intestinal crypts.61,62 This close association of the enteric glia cells and the intestinal crypt also points toward the formation of a spatially organized niche critical for intestine homeostasis.