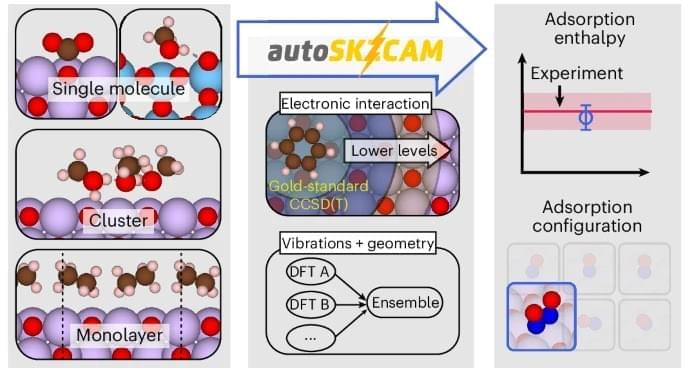
Understanding and predicting chemical reactions on surfaces lies at the heart of modern materials science. From heterogeneous catalysis to energy storage and greenhouse gas sequestration, surface chemistry defines the efficiency and viability of advanced technologies. Yet, computationally modeling these processes with both accuracy and efficiency has been a grand challenge.
A recent study published in Nature Chemistry introduces a breakthrough: the autoSKZCAM framework, an automated and open-source method that applies correlated wavefunction theory (cWFT) to surfaces of ionic materials at costs comparable to density functional theory (DFT). This achievement not only bridges the accuracy gap but also enables routine, large-scale studies of surface processes with chemical accuracy.