{kind=link}
Gene editing technologies—such as those used in agriculture and de-extinction projects—can be repurposed to offer what an international team of scientists is calling a transformative solution for restoring genetic diversity and saving endangered species.
Get the latest international news and world events from around the world.

Iron oxide behavior under pressure may reduce reliance on rare-earth metals in consumer, energy and medical tech
Researchers at The University of Texas at Arlington have discovered a surprising new type of magnetic property that could lead to stronger magnets made from tiny particles of common iron oxide. This finding could enhance the performance of everyday technologies while reducing the need for rare-earth metals—materials that are more costly, less sustainable and harder to obtain.
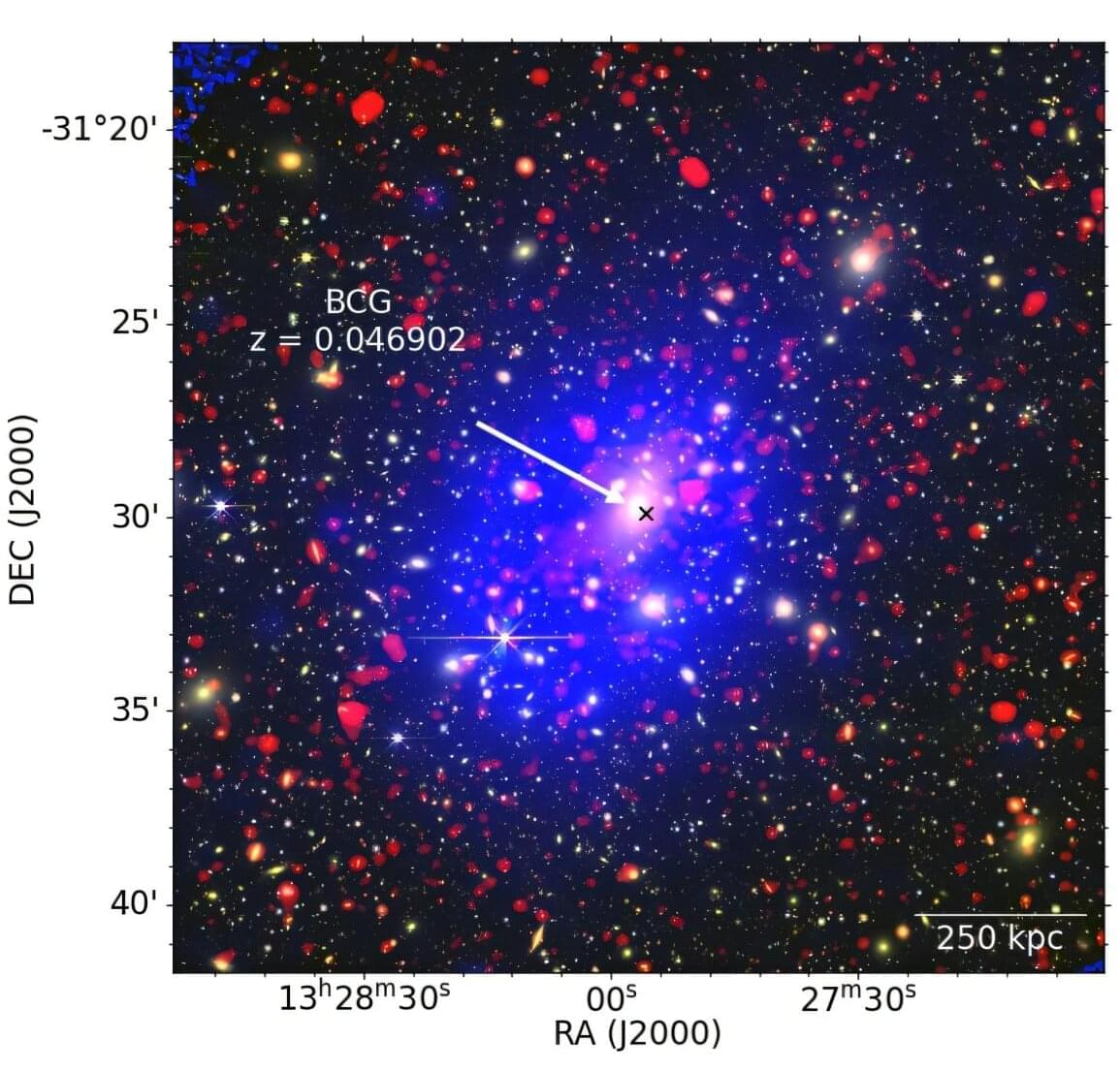
Galaxy cluster Abell 3558 has a peculiar mini-halo, observations suggest
An international team of astronomers has performed multi-band radio observations of diffuse radio emission in a galaxy cluster known as Abell 3558. As a result, the observational campaign detected that the cluster hosts a peculiar mini-halo. The finding was detailed in a paper published July 10 on the arXiv preprint server.
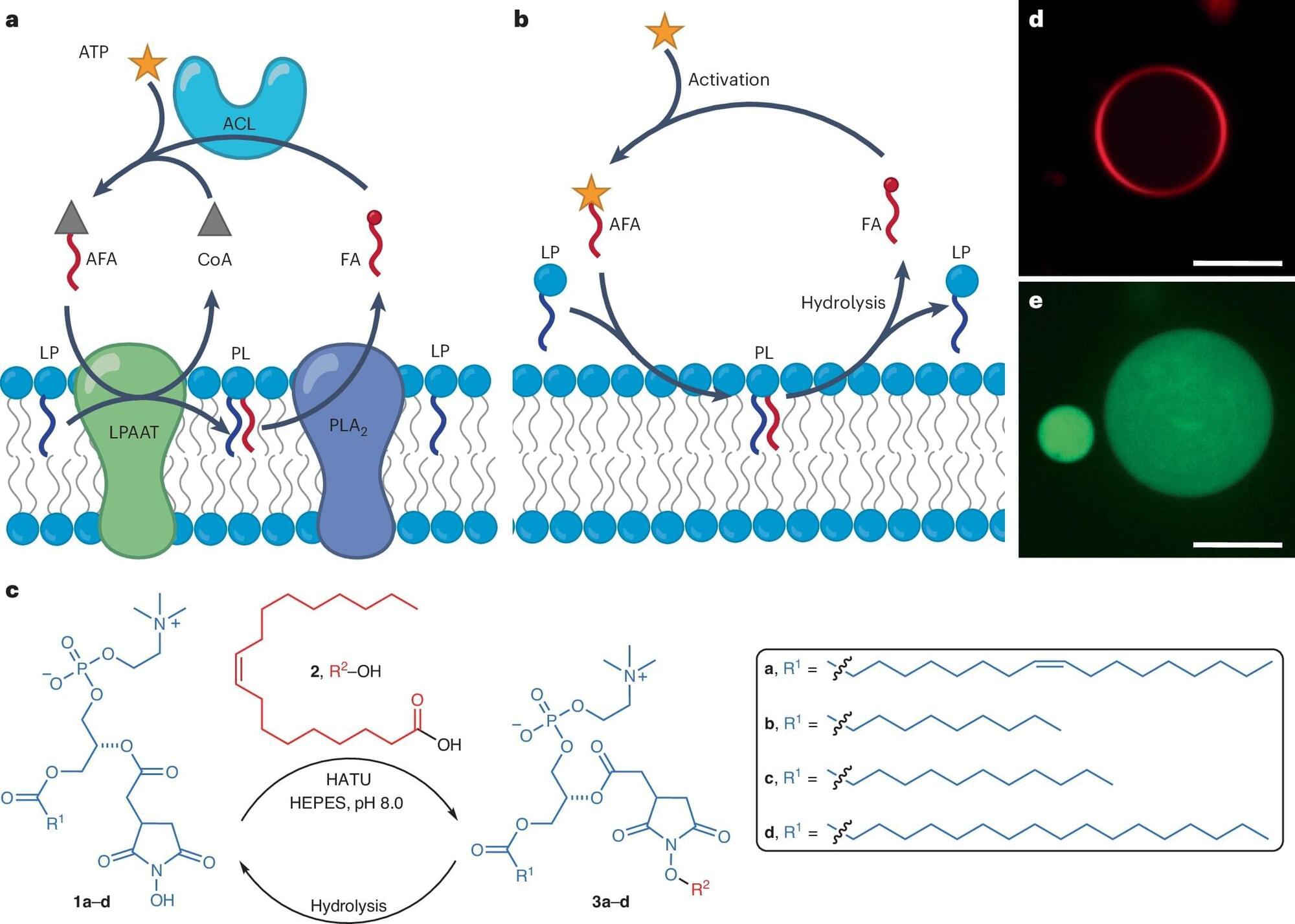
On the origins of life: Metabolic activity successfully incorporated into synthetic cell membranes
At some point during the evolution of life on Earth, inorganic matter became organic, nonliving matter became living. How this happened is one of humankind’s greatest mysteries. Today, scientists work to develop synthetic cells that mimic living cells, hoping to uncover clues that will help answer the question: how did life on Earth begin?
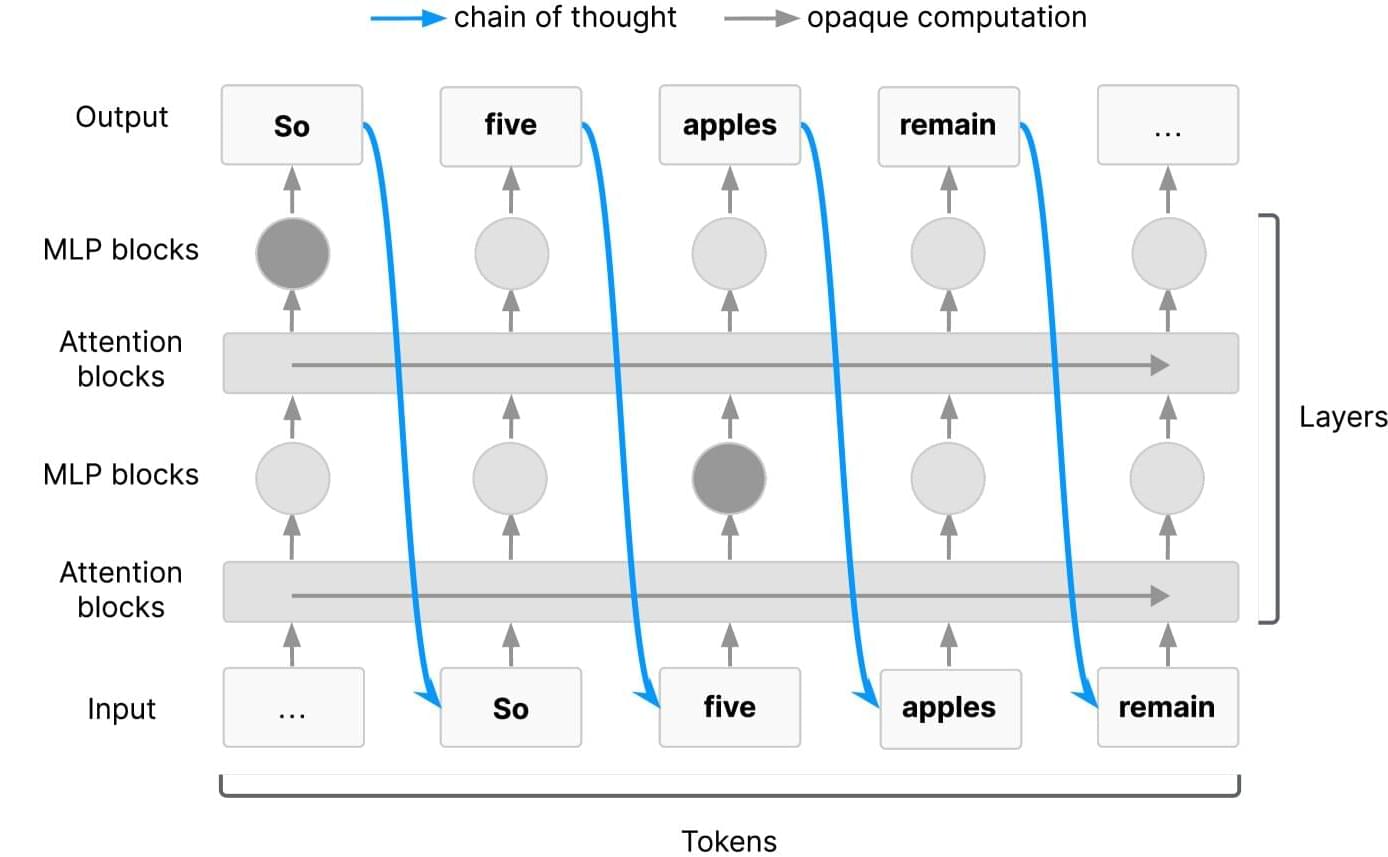
Tech giants warn window to monitor AI reasoning is closing, urge action
Artificial intelligence is advancing at a dizzying speed. Like many new technologies, it offers significant benefits but also poses safety risks. Recognizing the potential dangers, leading researchers from Google DeepMind, OpenAI, Meta, Anthropic and a coalition of companies and nonprofit groups have come together to call for more to be done to monitor how AI systems “think.”
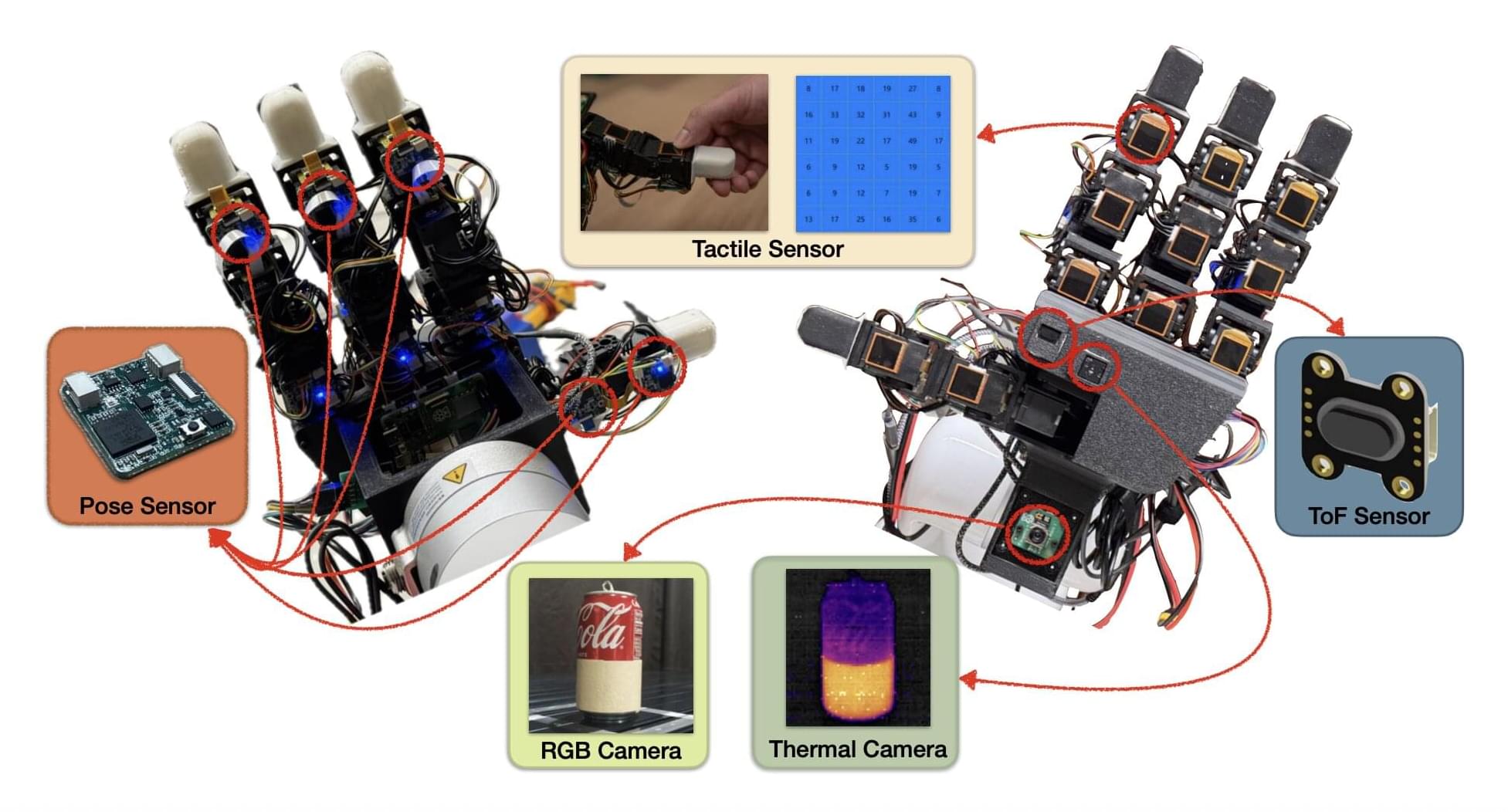
Dexterous robotic hand integrates thermal, inertial and force sensors
While roboticists have introduced increasingly advanced systems over the past decades, most existing robots are not yet able to manipulate objects with the same dexterity and sensing ability as humans. This, in turn, adversely impacts their performance in various real-world tasks, ranging from household chores to the clearing of rubble after natural disasters and the assembly or performing maintenance tasks, particularly in high-temperature working environments such as steel mills and foundries, where elevated temperatures can significantly degrade performance and compromise the precision required for safe operations.
Researchers at the University of Southern California recently developed the MOTIF (Multimodal Observation with Thermal, Inertial, and Force sensors) hand, a new robotic hand that could improve the object manipulation capabilities of humanoid robots. The innovation, presented in a paper posted to the arXiv preprint server, features a combination of sensing devices, including tactile sensors, a depth sensor, a thermal camera, inertial measurement unit (IMU) sensors and a visual sensor.
“Our paper emerged from the need to advance robotic manipulation beyond traditional visual and tactile sensing,” Daniel Seita, Hanyang Zhou, Wenhao Liu, and Haozhe Lou told Tech Xplore. “Current multi-fingered robotic hands often lack the integrated sensing capabilities necessary for complex tasks involving thermal awareness and responsive contact feedback.”
New study tackles dynamics of common—and difficult—sailing maneuver
Tacking—a maneuver used to sail a boat against the wind, changing direction in a zig-zag fashion—is one of the most difficult but necessary sailing maneuvers. While tacking is common, the movement of the sails and wind forces during the turn are not well understood.
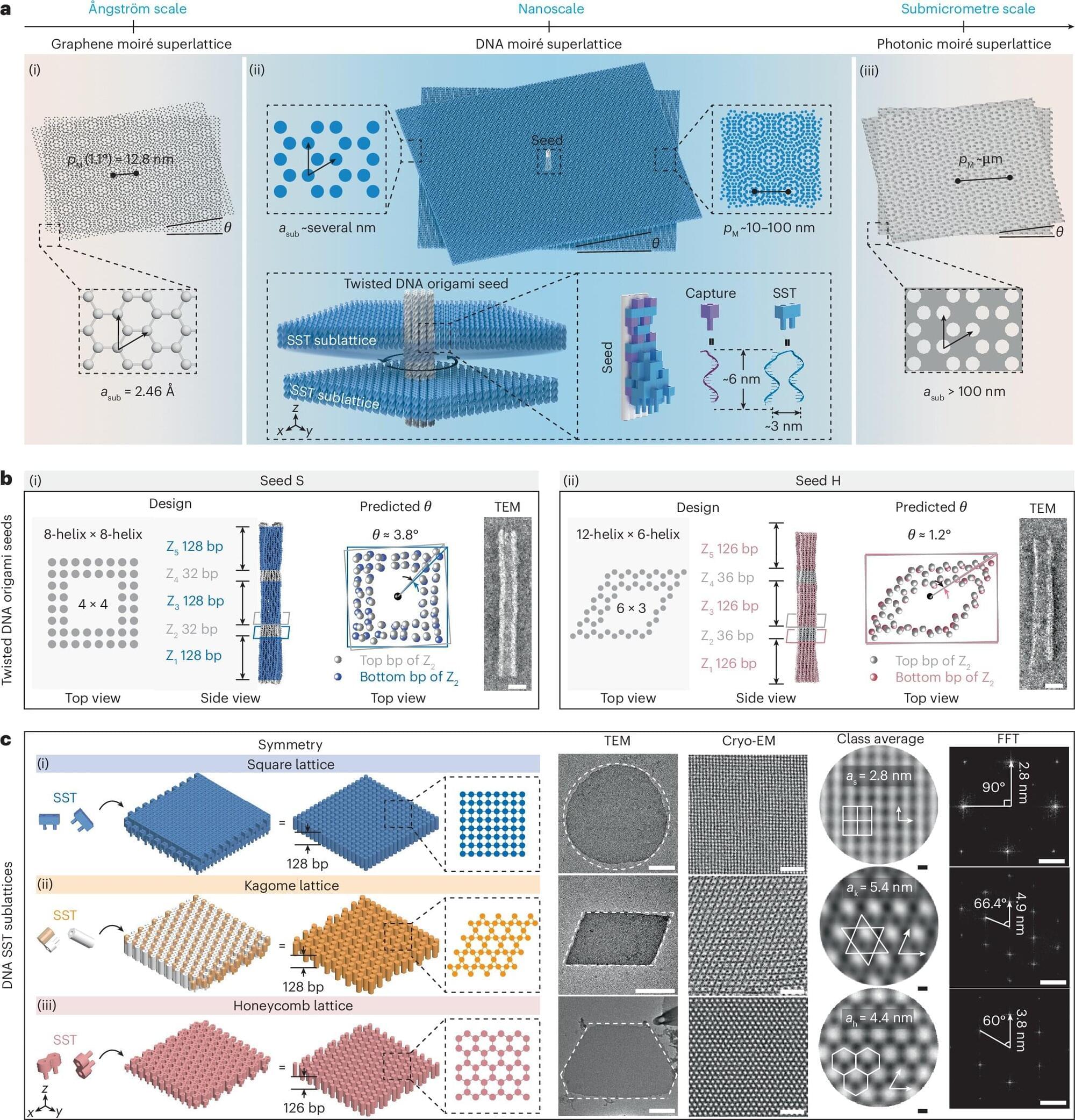
Expanding the material design space at the nanoscale
Researchers are creating new moiré materials at the nanometer scale using advanced DNA nanotechnology. DNA moiré superlattices form when two periodic DNA lattices are overlaid with a slight rotational twist or positional offset. This creates a new, larger interference pattern with completely different physical properties.

Can AI really code? Study maps the roadblocks to autonomous software engineering
Imagine a future where artificial intelligence quietly shoulders the drudgery of software development: refactoring tangled code, migrating legacy systems, and hunting down race conditions, so that human engineers can devote themselves to architecture, design, and the genuinely novel problems still beyond a machine’s reach.
Recent advances appear to have nudged that future tantalizingly close, but a new paper by researchers at MIT’s Computer Science and Artificial Intelligence Laboratory (CSAIL) and several collaborating institutions argues that this potential future reality demands a hard look at present-day challenges.
Titled “Challenges and Paths Towards AI for Software Engineering,” the work maps the many software-engineering tasks beyond code generation, identifies current bottlenecks, and highlights research directions to overcome them, aiming to let humans focus on high-level design while routine work is automated. The paper is available on the arXiv preprint server, and the researchers are presenting their work at the International Conference on Machine Learning (ICML 2025) in Vancouver.
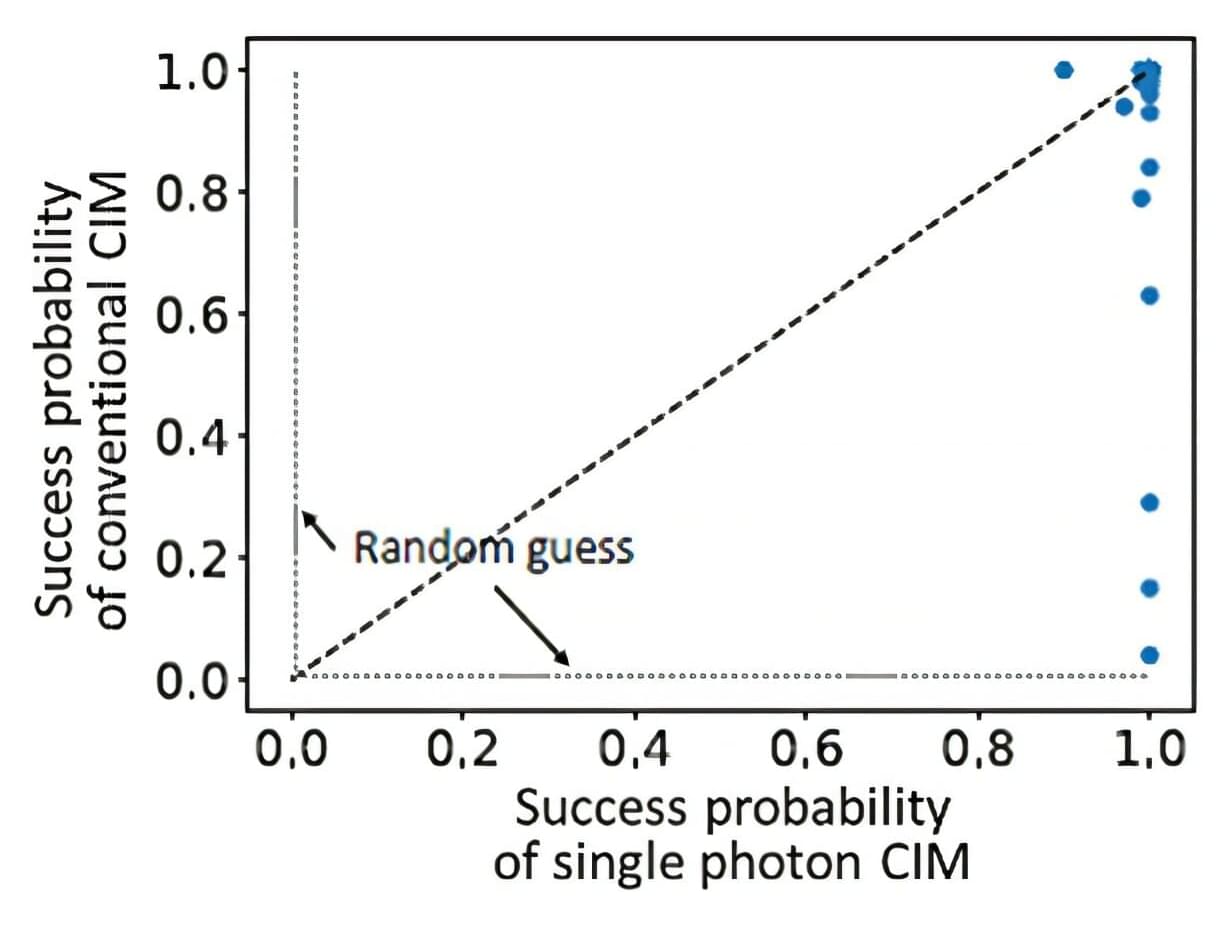
Toward quantum enhanced coherent Ising machines
The Graduate School of Information Science (GSIS) at Tohoku University, together with the Physics and Informatics (PHI) Lab at NTT Research, Inc., have jointly published a paper in the journal Quantum Science and Technology. The study involved studying a combinatorial clustering problem, a representative task in unsupervised machine learning.
Together, the two institutions are researching methods to bring to life a large-scale CIM simulation platform using conventional high-performance computing (HPC). This large-scale CIM will be critical to enabling cyber CIMs that will be widely accessible for solving hard NP, NP-complete and NP-hard problems.
The collaboration kicked off in 2023 with Hiroaki Kobayashi, Professor at the GSIS at Tohoku University, acting as the principal investigator for the joint research agreement (JRA), with PHI Lab Director Yoshihisa Yamamoto joining as the NTT Research counterpart to Kobayashi.