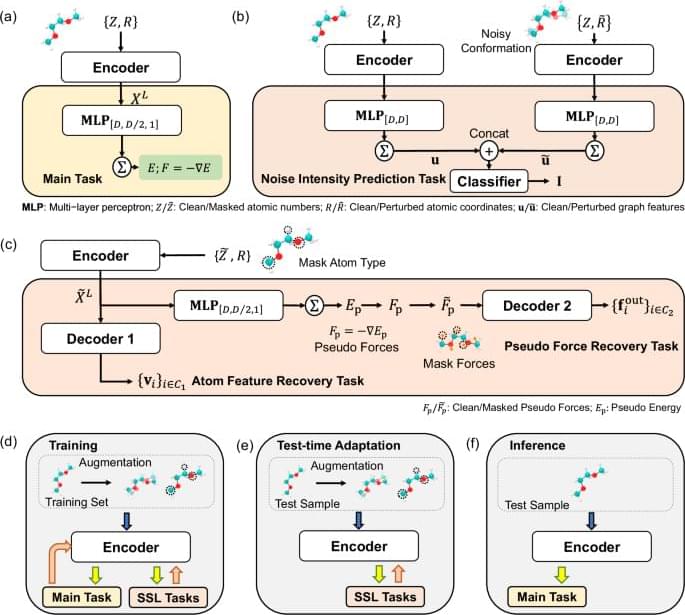
Molecular Dynamics (MD) simulation serves as a crucial technique across various disciplines including biology, chemistry, and material science1,2,3,4. MD simulations are typically based on interatomic potential functions that characterize the potential energy surface of the system, with atomic forces derived as the negative gradients of the potential energies. Subsequently, Newton’s laws of motion are applied to simulate the dynamic trajectories of the atoms. In ab initio MD simulations5, the energies and forces are accurately determined by solving the equations in quantum mechanics. However, the computational demands of ab initio MD limit its practicality in many scenarios. By learning from ab initio calculations, machine learning interatomic potentials (MLIPs) have been developed to achieve much more efficient MD simulations with ab initio-level accuracy6,7,8.
Despite their successes, the crucial challenge of implementing MLIPs is the distribution shift between training and test data. When using MLIPs for MD simulations, the data for inference are atomic structures that are continuously generated during simulations based on the predicted forces, and the training set should encompass a wide range of atomic structures to guarantee the accuracy of predictions. However, in fields such as phaseion9,10, catalysis11,12, and crystal growth13,14, the configurational space that needs to be explored is highly complex. This complexity makes it challenging to sample sufficient data for training and easy to make a potential that is not smooth enough to extrapolate to every relevant point. Consequently, a distribution shift between training and test datasets often occurs, which causes the degradation of test performance and leads to the emergence of unrealistic atomic structures, and finally the MD simulations collapse15.