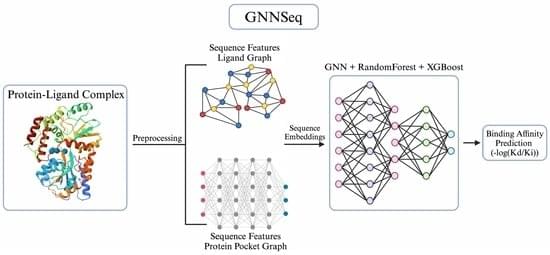
Background/Objectives: Accurately predicting protein–ligand binding affinity is essential in drug discovery for identifying effective compounds. While existing sequence-based machine learning models for binding affinity prediction have shown potential, they lack accuracy and robustness in pattern recognition, which limits their generalizability across diverse and novel binding complexes. To overcome these limitations, we developed GNNSeq, a novel hybrid machine learning model that integrates a Graph Neural Network (GNN) with Random Forest (RF) and XGBoost. Methods: GNNSeq predicts ligand binding affinity by extracting molecular characteristics and sequence patterns from protein and ligand sequences. The fully optimized GNNSeq model was trained and tested on subsets of the PDBbind dataset. The novelty of GNNSeq lies in its exclusive reliance on sequence features, a hybrid GNN framework, and an optimized kernel-based context-switching design. By relying exclusively on sequence features, GNNSeq eliminates the need for pre-docked complexes or high-quality structural data, allowing for accurate binding affinity predictions even when interaction-based or structural information is unavailable. The integration of GNN, XGBoost, and RF improves GNNSeq performance by hierarchical sequence learning, handling complex feature interactions, reducing variance, and forming a robust ensemble that improves predictions and mitigates overfitting. The GNNSeq unique kernel-based context switching scheme optimizes model efficiency and runtime, dynamically adjusts feature weighting between sequence and basic structural information, and improves predictive accuracy and model generalization. Results: In benchmarking, GNNSeq performed comparably to several existing sequence-based models and achieved a Pearson correlation coefficient (PCC) of 0.784 on the PDBbind v.2020 refined set and 0.84 on the PDBbind v.2016 core set. During external validation with the DUDE-Z v.2023.06.20 dataset, GNNSeq attained an average area under the curve (AUC) of 0.74, demonstrating its ability to distinguish active ligands from decoys across diverse ligand–receptor pairs. To further evaluate its performance, we combined GNNSeq with two additional specialized models that integrate structural and protein–ligand interaction features. When tested on a curated set of well-characterized drug–target complexes, the hybrid models achieved an average PCC of 0.89, with the top-performing model reaching a PCC of 0.97. GNNSeq was designed with a strong emphasis on computational efficiency, training on 5000+ complexes in 1 h and 32 min, with real-time affinity predictions for test complexes. Conclusions: GNNSeq provides an efficient and scalable approach for binding affinity prediction, offering improved accuracy and generalizability while enabling large-scale virtual screening and cost-effective hit identification. GNNSeq is publicly available in a server-based graphical user interface (GUI) format.