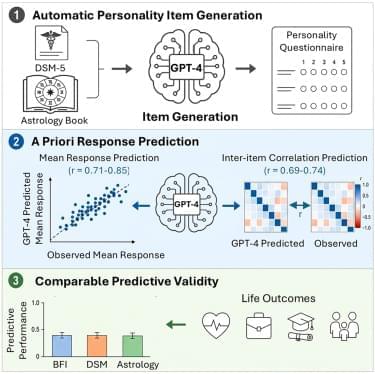
The convergence of AI with robotics, biotechnology, neuromorphic computing, and brain-computer interfaces is accelerating innovation, ushering in a “cyborg horizon.”

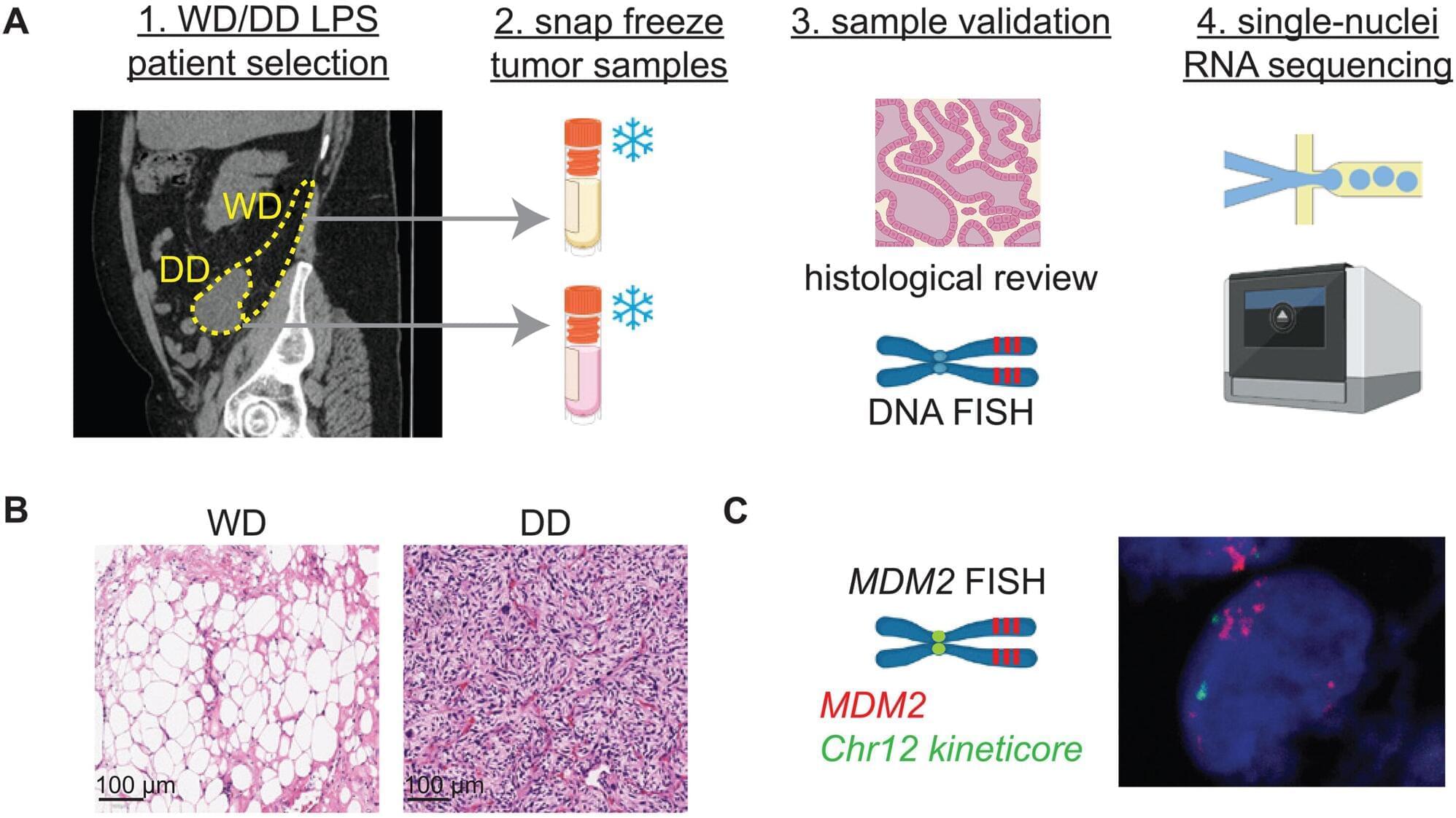
New research from an expert in cancer biology explains what triggers liposarcoma cells to become more or less aggressive and identifies potential targets that could keep aggressive tumors in check. A study led by Blake Wilde, Ph.D., of Roswell Park Comprehensive Cancer Center highlights how this discovery may pave the way for new treatment options for this cancer.
The findings are published in the journal Science Advances.
More than half of all people with liposarcoma will see their cancer return after treatment, underscoring the need for new and better treatment options. The two most common types of this cancer, which begins in fat tissue, are well-differentiated (WD) and dedifferentiated (DD) liposarcoma, explains the study’s first author, Blake Wilde, Ph.D., assistant professor of oncology in the Departments of Urology and Cell Stress Biology at Roswell Park.
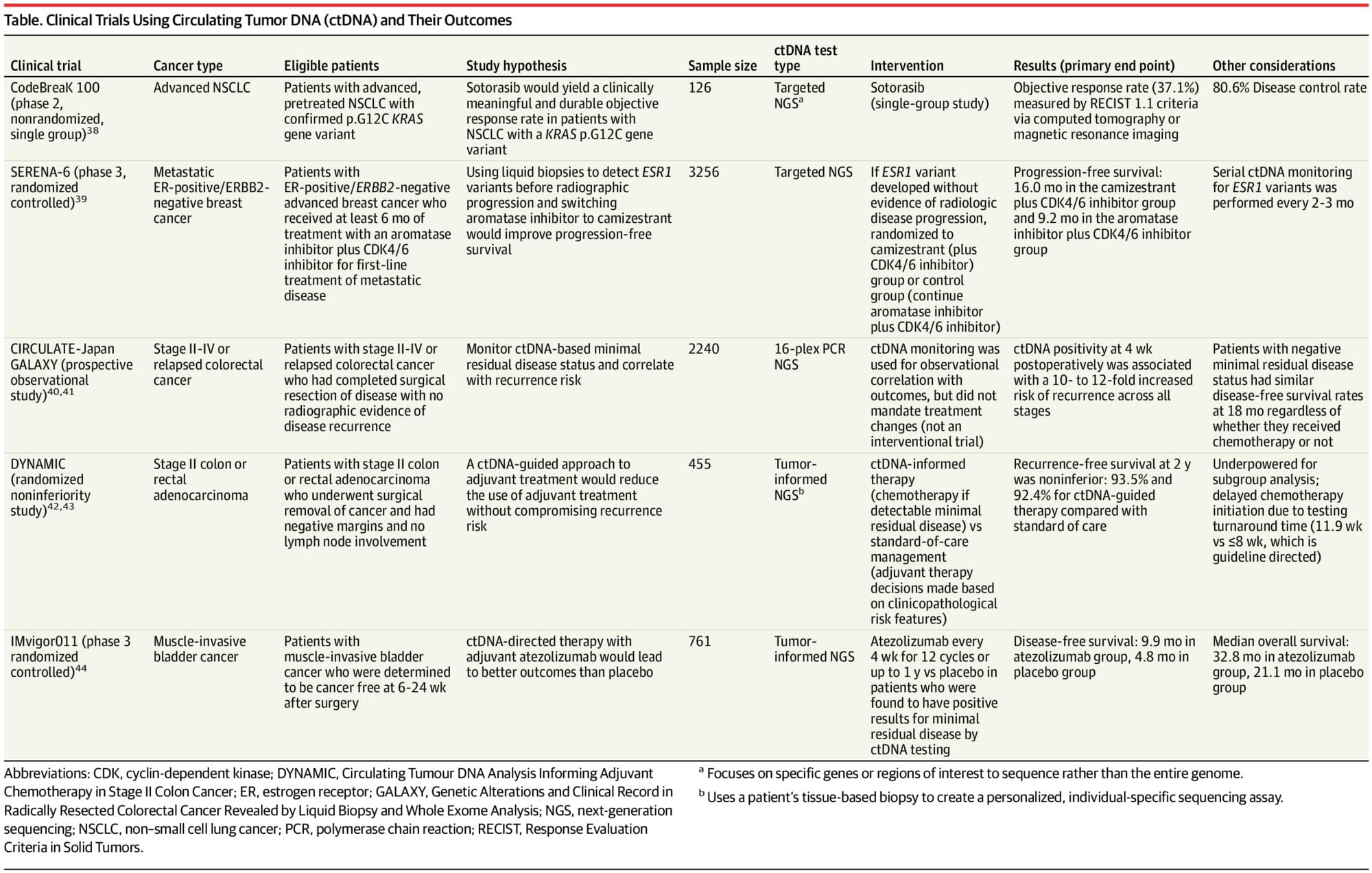
Three research teams working independently around the world have landed on the same discovery: A single molecular “handshake” inside our cells controls a family of inflammatory diseases, including one of the most common inherited fevers on Earth. The finding solved a decades-old puzzle for one family and led to a treatment that worked almost immediately.
It all revolves around Familial Mediterranean Fever (FMF), the most common inherited autoinflammatory disease, which affects an estimated 1–2 in every 1,000 people in high-prevalence populations, including those of Mediterranean, Middle Eastern, Armenian and Jewish ancestry. FMF begins in childhood, causing recurring fevers, painful rashes and joint pain.
For more than 20 years, one family lived with a mysterious illness that resembled FMF, but nothing doctors tried would cure it.
2 Department of Breast Surgery of the Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
3Department of Radiation Oncology, Zhejiang Cancer Hospital, Hangzhou Institute of Medicine, Chinese Academy of Sciences, Zhejiang Key Laboratory of Particle Radiotherapy Equipment, Hangzhou, China.


How our solar system moves through space. This is a non-conventional view of our solar system that is different from the standard ‘flat’ diagrams. We travel, never return to the same spot again. Helical motion, how the planets move through space.
Respect my copyright and do not re-upload.
Like the music? https://djsadhu.bandcamp.com/
Please consider becoming a Patron to support my work: / djsadhu.
Thanks to the community for providing translations!
New version: Solar System 2.0: • Solar System 2.0 — the helical model.
Full story and philosophy: http://www.djsadhu.com/research/solar… 2 is here: • The helical model — our Galaxy is a vortex Information & research: http://www.djsadhu.com/the-helical-mo… Music: https://djsadhu.bandcamp.com/album/dj… French subtitles provided by the Resonance Project Greek subtitles provided by vasoula2908 Download the sound track: http://www.djsadhu.com/audio-video/vo… (FOR PERSONAL USE ONLY) No, this was not made with Universe Sandbox, but with 3DsMax. Yes, I messed up two orbits.
PART 2 is here: • The helical model — our Galaxy is a vortex.
Information & research: http://www.djsadhu.com/the-helical-mo…
Music: https://djsadhu.bandcamp.com/album/dj…
French subtitles provided by the Resonance Project.
Greek subtitles provided by vasoula2908
Download the sound track: http://www.djsadhu.com/audio-video/vo… (FOR PERSONAL USE ONLY)
No, this was not made with Universe Sandbox, but with 3DsMax.
Yes, I messed up two orbits.
{kind=link}