Cryonics is one of the most misunderstood ideas in science today: most people think it means “freezing dead people.” It doesn’t. This video breaks down what…
Category: life extension
Premature blood stem cell aging in sickle cell disease may be reversible
Sickle cell disease causes premature aging of blood stem cells, which scientists may be able to address with a special class of drugs, according to a new study from St. Jude Children’s Research Hospital. Patients with sickle cell disease experience higher rates of blood stem cell dysfunction and blood cancers compared with their peers, though the reason has been unclear.
The St. Jude researchers found that blood stem cells from young patients with sickle cell disease have features of aging, likely increasing the risk of other complications. Giving senolytics, which target aging-related processes, improved disease symptoms in model systems, with implications for curative gene therapies. The findings were published in Science Translational Medicine.
“We saw that blood stem cells from even young patients with sickle cell disease have many markers of senescence or aging,” said senior co-corresponding author Shannon McKinney-Freeman, Ph.D., St. Jude Department of Hematology.
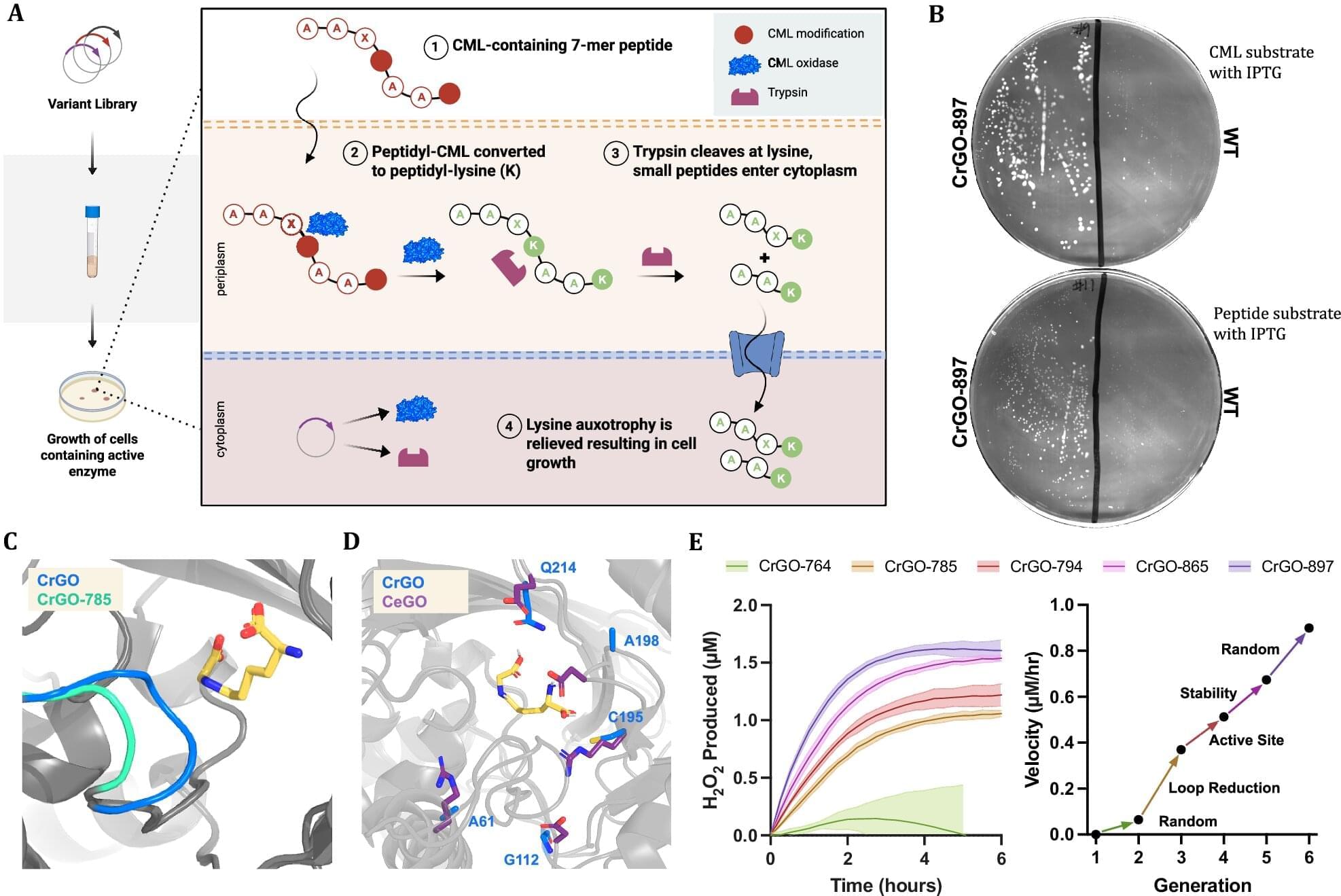
Engineered enzyme erases a stubborn mark of aging by up to 70% in human tissue samples
A biotech company called Revel Pharmaceuticals is looking into ways to reverse aging, and the company’s science team, along with researchers from the company Calico and the University of Colorado, may be a step closer to realizing the so-called fountain of youth. The team recently published their study in Nature Communications detailing how they engineered an enzyme capable of reversing a particular form of age-related damage and demonstrated its competence with test results.
One common sign of aging in the cells of living organisms is a type of protein damage called Nε-carboxymethyl-lysine (CML). CML is part of a group of harmful compounds aptly named “AGEs” (or advanced glycation and lipoxidation end products). Oddly enough, it is also part of the Maillard reaction, known for causing the browning in cooked food that creates rich, savory flavors, complex aromas and golden-brown crusts. In living organisms, CML builds up on long-lived proteins, like those in skin, blood vessels and the eye. This stiffens tissues and can fuel chronic inflammation through an immune-signaling receptor called “RAGE.”
“The engagement of the CML-RAGE axis triggers a signaling cascade that activates NF-κB and stimulates the release of pro-inflammatory cytokines and profibrotic growth factors. In the context of the central nervous system, CML accumulation has been linked to oxidative stress and mitochondrial damage in microglia, further disrupting brain homeostasis during aging,” the authors of the new study explain.
Ultimate Limit of The Human Lifespan May Be Identified, Study Suggests
{kind=link}
When the iconic rock band Queen asked, “Who wants to live forever?” the question was rhetorical, but for many people, the answer was “Yes”
Well, a new study suggests immortal life may be scientifically impossible, even if we somehow found the perfect anti-aging medicine.
If scientists managed to overcome every other aspect of aging, humans still couldn’t live forever, the new research shows. Random DNA mutations would continue accumulating in our cells until the body could no longer function.
Lifespan Extension Expert — AMA (pt.1)
Many people want to live longer than what is currently possible, but medical technology is not progressing fast enough. At Tomorrow Biostasis, we use the latest cryopreservation technology to medically preserve and protect you for as long as it is needed. When medical technology has solved the life extension and aging problems, you will be reanimated to enjoy an extended life.
On the YouTube channel of Tomorrow Biostasis you can find more information about the concepts of cryopreservation, cryonics, biostasis, vitrification, human revival, and more. We also provide you with practical information about signing up with Tomorrow Biostasis. Get ready to get an insight into the fascinating world of cryopreservation!
Visit our website at: https://bit.ly/35KsOIq.
Sign up for Biostasis: https://bit.ly/3hGb6sq.
Schedule a free Consultation: https://bit.ly/3IIwkll.
Also follow Tomorrow Biostasis on
Twitter: https://bit.ly/374bQ8z.
Instagram: https://bit.ly/3HFMpqr.
Facebook: https://bit.ly/3tt9bg3
LinkedIn: https://bit.ly/3hEtjX7
Reddit: https://bit.ly/3CeE8bY
Discord: / discord.
#TomorrowBiostasis #Cryopreservation #Cryonics.
Tomorrow Biostasis | © Copyright 2021–2022
AI And The Future Of Healthspan
Join us on Patreon! / michaellustgartenphd.
Discount Links/Affiliates:
Blood testing (where I get the majority of my labs, for those who blood test with Quest): https://www.ultalabtests.com/partners… those who blood test with LabCorp: https://www.anrdoezrs.net/click-10161… At-Home Metabolomics: https://www.iollo.com?ref=michael-lus… Use Code: CONQUERAGING At Checkout Clearly Filtered Water Filter: https://get.aspr.app/SHoPY Epigenetic, Telomere Testing: https://trudiagnostic.com/?irclickid=… Use Code: CONQUERAGING NAD+ Quantification: https://www.jinfiniti.com/intracellul… Use Code: ConquerAging At Checkout Oral Microbiome: https://www.bristlehealth.com/?ref=mi… Enter Code: ConquerAging SiphoxHealth Blood Testing (ApoB, GrimAge): https://siphoxhealth.com/mlustgarten Green Tea: https://www.ochaandco.com/?ref=fqbtflod Use Code: ML10OFF Diet Tracking: https://shareasale.com/r.cfm?b=139013… If you’d like to support the channel, you can do that with the website, Buy Me A Coffee: https://www.buymeacoffee.com/mlhnrca Conquer Aging Or Die Trying Merch! https://my-store-d4e7df.creator-sprin…
Blood Testing Essentials (Biological Age, CVD-Risk, Kidney Health and Function):
PhenoAge (Biological Age): https://www.ultalabtests.com/partners…
Measure the Bortz biological clock biomarkers: https://www.ultalabtests.com/partners…
Calculate your biological age using the Bortz clock: https://www.longevity-tools.com/human…